The Autocrine FGF/FGFR System in both Skin and Uveal Melanoma: FGF Trapping as a Possible Therapeutic Approach

 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
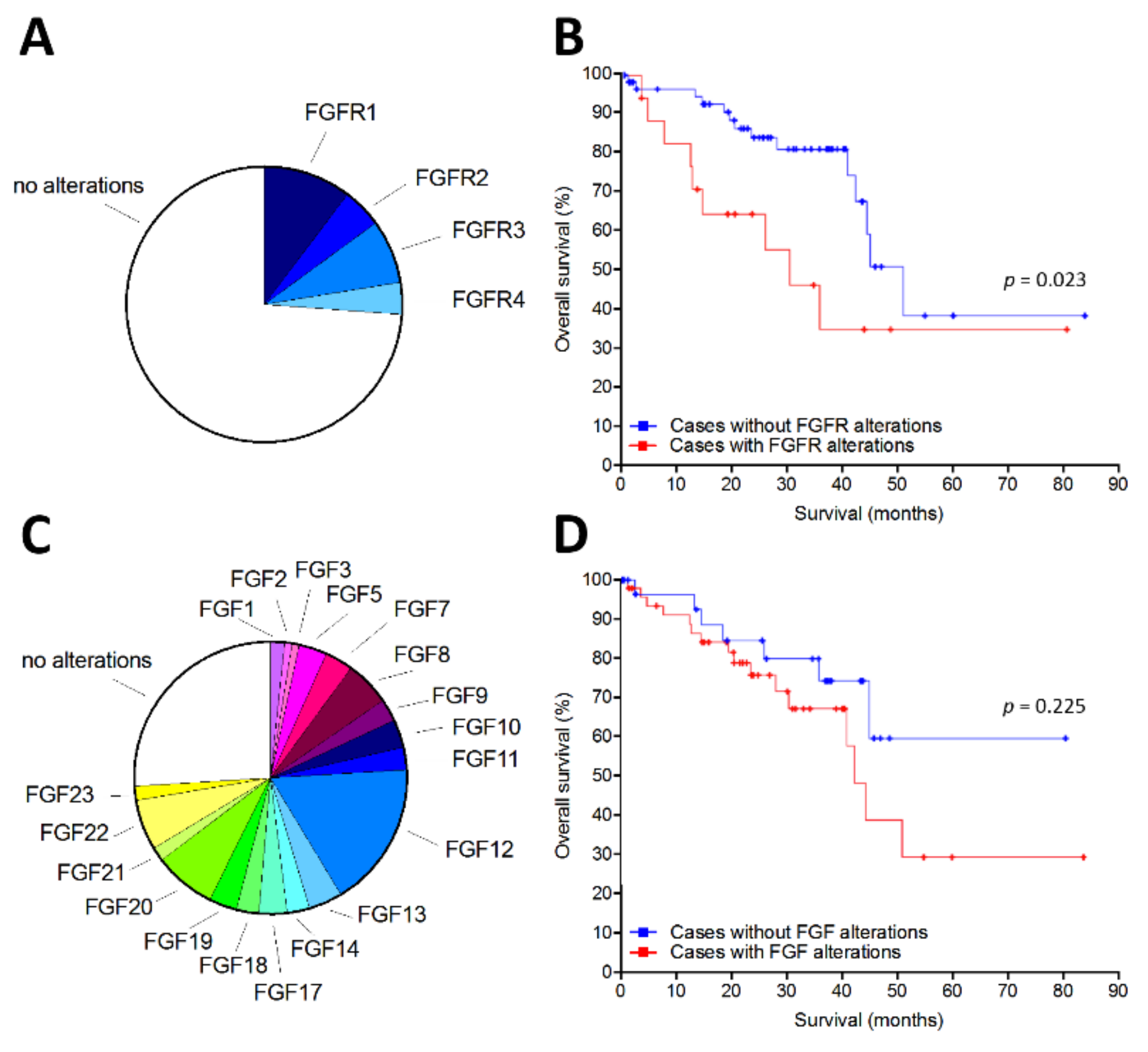
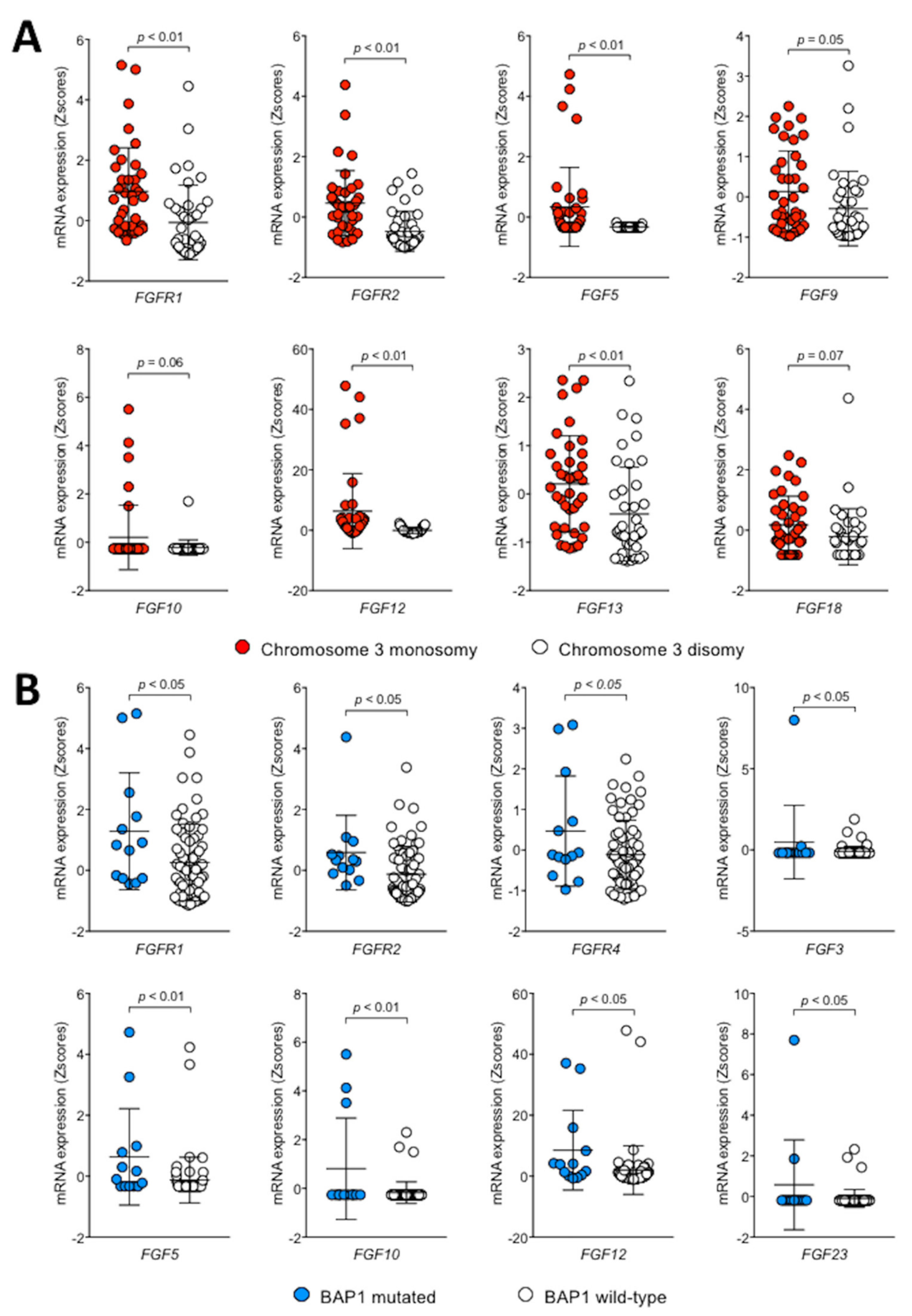
2.1. The FGF/FGFR System Is Upregulated in Human Uveal Melanoma
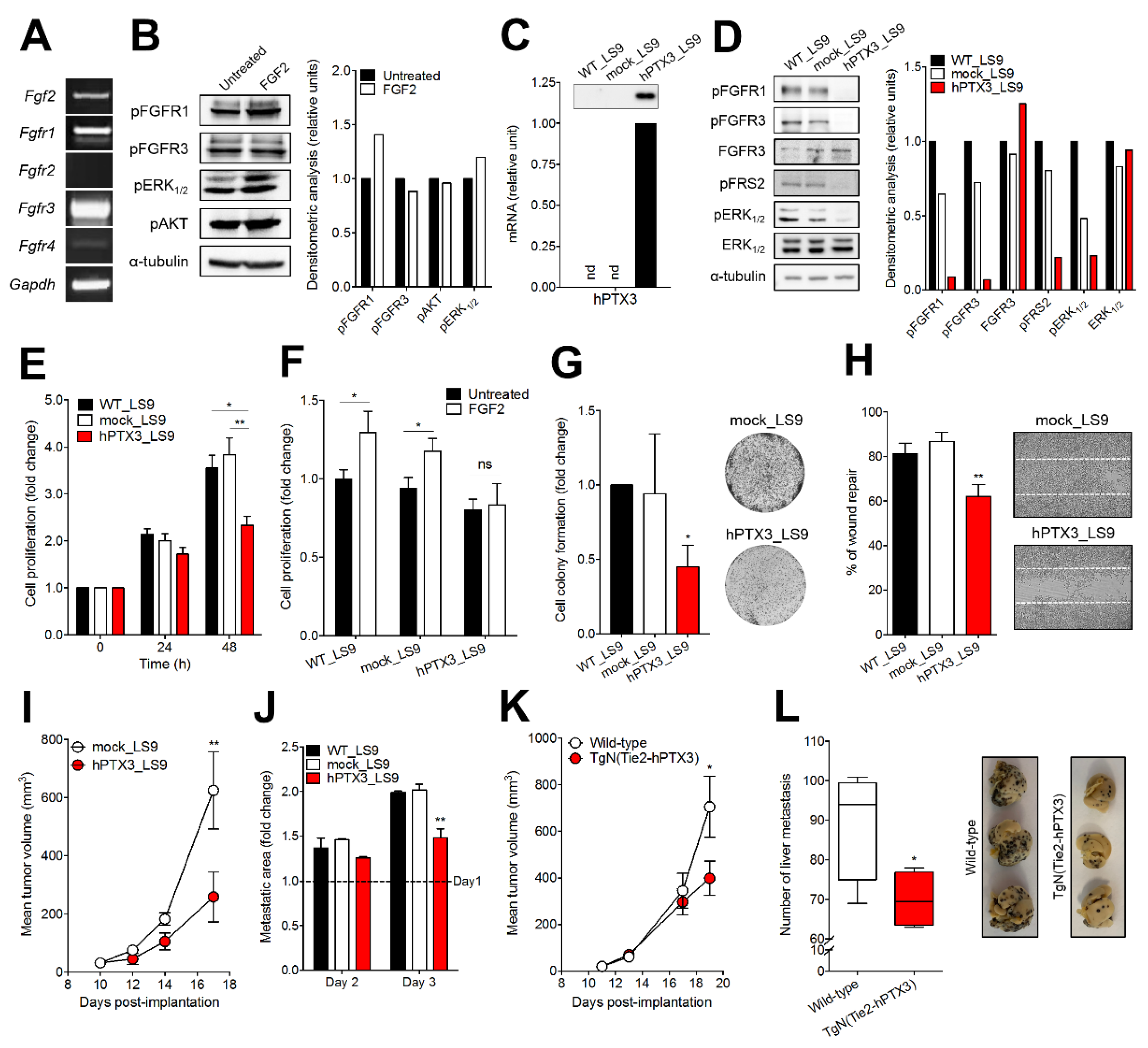
2.2. PTX3 Inhibits the Tumorigenic and Metastatic Activity of Murine B16-LS9 Cells
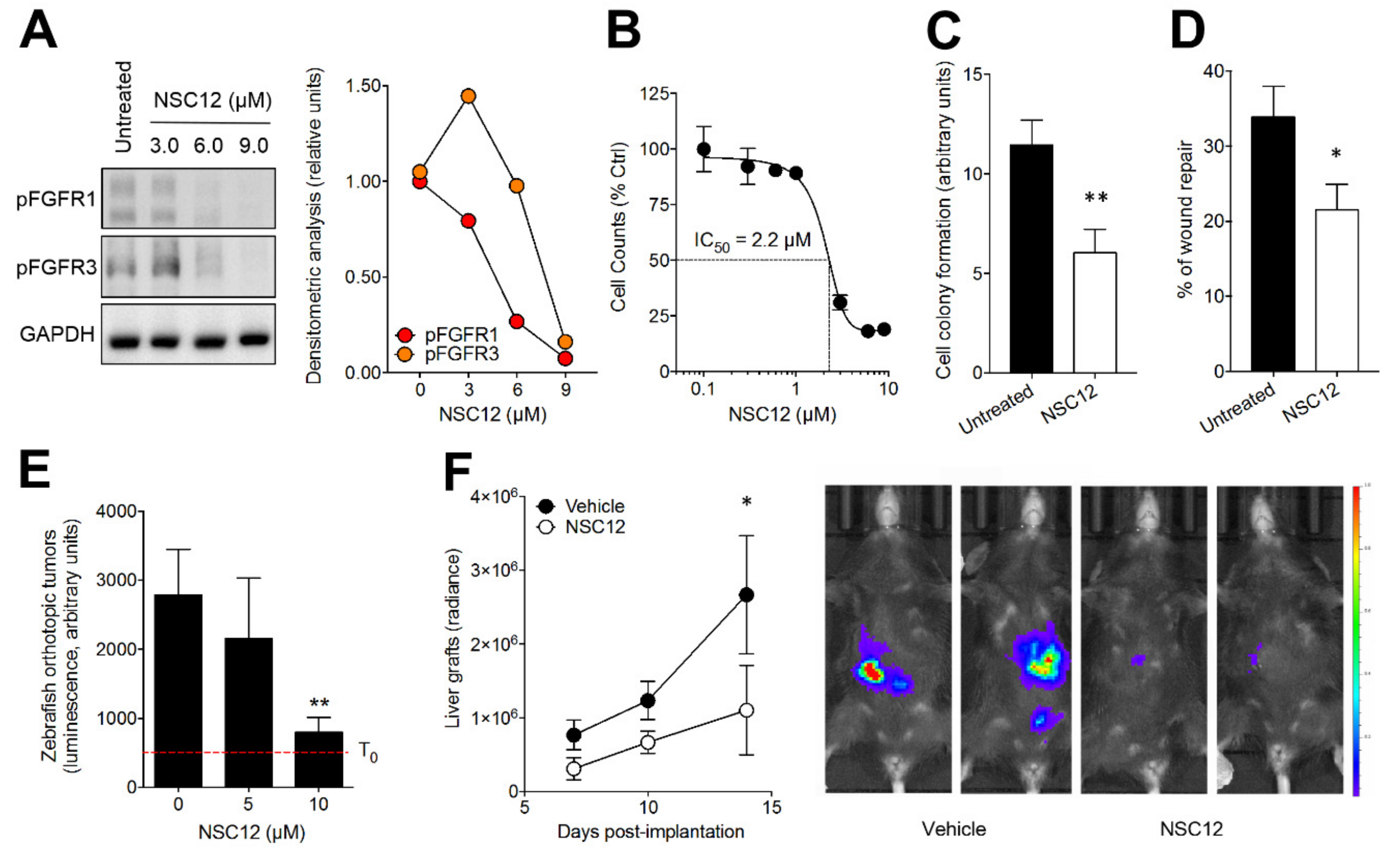
2.3. The Pan-FGF Trap NSC12 Inhibits the Tumorigenic and Metastatic Activity of Murine B16-LS9 Cells
2.4. FGF Trapping Inhibits Human FGF/FGFR Signaling and Proliferation in UM Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures
4.3. Real-Time PCR Analysis
4.4. Western Blot Analysis
4.5. Cell Proliferation Assay
4.6. Colony Formation Assay
4.7. Chemotaxis Assay
4.8. Wound Healing Assay
4.9. Apoptotic Cell Death Analysis
4.10. Tumor Graft and Liver Metastasis Assays in Mice
4.11. Zebrafish Embryo Assays
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jovanovic, P.; Mihajlovic, M.; Djordjevic-Jocic, J.; Vlajkovic, S.; Cekic, S.; Stefanovic, V. Ocular melanoma: an overview of the current status. Int. J. Clin. Exp. Pathol. 2013, 6, 1230–1244. [Google Scholar] [PubMed]
- Mahendraraj, K.; Lau, C.S.; Lee, I.; Chamberlain, R.S. Trends in incidence, survival, and management of uveal melanoma: a population-based study of 7,516 patients from the Surveillance, Epidemiology, and End Results database (1973-2012). Clin. Ophthalmol. 2016, 10, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, Y.; Kim, I.K. Epidemiology and management of uveal melanoma. Hematol. Oncol. Clin. North Am. 2012, 26, 1169–1184. [Google Scholar] [CrossRef] [PubMed]
- Vivet-Noguer, R.; Tarin, M.; Roman-Roman, S.; Alsafadi, S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Mathis, T.; Cassoux, N.; Tardy, M.; Piperno, S.; Gastaud, L.; Dendale, R.; Maschi, C.; Nguyen, A.M.; Meyer, L.; Bonnin, N.; et al. Management of uveal melanomas, guidelines for oncologists. Bull. Cancer 2018, 105, 967–980. [Google Scholar] [CrossRef]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert. Rev. Anticancer Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR system as a "two-compartment" antiangiogenic/antitumor approach in cancer therapy. Pharmacol. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef]
- Boyd, S.R.; Tan, D.S.; de Souza, L.; Neale, M.H.; Myatt, N.E.; Alexander, R.A.; Robb, M.; Hungerford, J.L.; Cree, I.A. Uveal melanomas express vascular endothelial growth factor and basic fibroblast growth factor and support endothelial cell growth. Br. J. Ophthalmol. 2002, 86, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Notting, I.C.; Missotten, G.S.; Sijmons, B.; Boonman, Z.F.; Keunen, J.E.; van der Pluijm, G. Angiogenic profile of uveal melanoma. Curr. Eye Res. 2006, 31, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, G.; Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.M.; Mrzyk, S.; Mascarelli, F. Activation of the FGF2/FGFR1 autocrine loop for cell proliferation and survival in uveal melanoma cells. Invest. Ophthalmol. Vis. Sci. 2009, 50, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bao, X.; Zhang, Z.; Sun, Y.; Zhou, X. FGF2 promotes metastasis of uveal melanoma cells via store-operated calcium entry. Onco. Targets Ther. 2017, 10, 5317–5328. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Bottazzi, B.; Bastone, A.; Mantovani, A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu. Rev. Immunol. 2005, 23, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, A.; Ghedini, G.C.; Presta, M.; Ronca, R. Long pentraxin 3: A novel multifaceted player in cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Leali, D.; Alessi, P.; Coltrini, D.; Ronca, R.; Corsini, M.; Nardo, G.; Indraccolo, S.; Presta, M. Long pentraxin-3 inhibits FGF8b-dependent angiogenesis and growth of steroid hormone-regulated tumors. Mol. Cancer Ther. 2011, 10, 1600–1610. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Alessi, P.; Coltrini, D.; Di Salle, E.; Giacomini, A.; Leali, D.; Corsini, M.; Belleri, M.; Tobia, C.; Garlanda, C.; et al. Long pentraxin-3 as an epithelial-stromal fibroblast growth factor-targeting inhibitor in prostate cancer. J. Pathol. 2013, 230, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, M.; Rusnati, M.; Bugatti, A.; Bottazzi, B.; Mantovani, A.; Bastone, A.; Inforzato, A.; Vincenti, S.; Bracci, L.; Mastroianni, D.; et al. Identification of an antiangiogenic FGF2-binding site in the N terminus of the soluble pattern recognition receptor PTX3. J. Biol. Chem. 2006, 281, 22605–22613. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Di Salle, E.; Giacomini, A.; Leali, D.; Alessi, P.; Coltrini, D.; Ravelli, C.; Matarazzo, S.; Ribatti, D.; Vermi, W.; et al. Long pentraxin-3 inhibits epithelial-mesenchymal transition in melanoma cells. Mol. Cancer Ther. 2013, 12, 2760–2771. [Google Scholar] [CrossRef]
- Castelli, R.; Giacomini, A.; Anselmi, M.; Bozza, N.; Vacondio, F.; Rivara, S.; Matarazzo, S.; Presta, M.; Mor, M.; Ronca, R. Synthesis, Structural Elucidation, and Biological Evaluation of NSC12, an Orally Available Fibroblast Growth Factor (FGF) Ligand Trap for the Treatment of FGF-Dependent Lung Tumors. J. Med. Chem. 2016, 59, 4651–4663. [Google Scholar] [CrossRef] [Green Version]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torrella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, F.; De Smet, F.; Herbert, C.; De Bock, K.; Georgiadou, M.; Fons, P.; Tjwa, M.; Alcouffe, C.; Ny, A.; Bianciotto, M.; et al. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell 2013, 23, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220 e215. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, N.; Thomton, S.; Coupland, S.E.; Coulson, J.M.; Sacco, J.J.; Krishna, Y.; Heimann, H.; Taktak, A.; Cebulla, C.M.; Abdel-Rahman, M.H.; et al. Patterns of BAP1 protein expression provide insights into prognostic significance and the biology of uveal melanoma. J. Path. Clin. Res. 2018, 4, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Diaz, C.E.; Rusciano, D.; Dithmar, S.; Grossniklaus, H.E. B16LS9 melanoma cells spread to the liver from the murine ocular posterior compartment (PC). Curr. Eye Res. 1999, 18, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Rusciano, D.; Lorenzoni, P.; Burger, M.M. Regulation of c-met expression in B16 murine melanoma cells by melanocyte stimulating hormone. J. Cell Sci. 1999, 112, 623–630. [Google Scholar] [PubMed]
- Elia, G.; Ren, Y.; Lorenzoni, P.; Zarnegar, R.; Burger, M.M.; Rusciano, D. Mechanisms regulating c-met overexpression in liver-metastatic B16-LS9 melanoma cells. J. Cell Biochem. 2001, 81, 477–487. [Google Scholar] [CrossRef]
- Jones, N.M.; Yang, H.; Zhang, Q.; Morales-Tirado, V.M.; Grossniklaus, H.E. Natural killer cells and pigment epithelial-derived factor control the infiltrative and nodular growth of hepatic metastases in an Orthotopic murine model of ocular melanoma. BMC Cancer 2019, 19, 484. [Google Scholar] [CrossRef]
- Yang, W.; Li, H.; Mayhew, E.; Mellon, J.; Chen, P.W.; Niederkorn, J.Y. NKT cell exacerbation of liver metastases arising from melanomas transplanted into either the eyes or spleens of mice. Invest. Ophthalmol. Vis. Sci. 2011, 52, 3094–3102. [Google Scholar] [CrossRef]
- Yang, H.; Brackett, C.M.; Morales-Tirado, V.M.; Li, Z.; Zhang, Q.; Wilson, M.W.; Benjamin, C.; Harris, W.; Waller, E.K.; Gudkov, A.V.; et al. The Toll-like receptor 5 agonist entolimod suppresses hepatic metastases in a murine model of ocular melanoma via an NK cell-dependent mechanism. Oncotarget 2016, 7, 2936–2950. [Google Scholar] [CrossRef]
- Ashur-Fabian, O.; Zloto, O.; Fabian, I.; Tsarfaty, G.; Ellis, M.; Steinberg, D.M.; Hercbergs, A.; Davis, P.J.; Fabian, I.D. Tetrac Delayed the Onset of Ocular Melanoma in an Orthotopic Mouse Model. Front. Endocrinol. 2018, 9, 775. [Google Scholar] [CrossRef] [PubMed]
- Tobia, C.; Gariano, G.; De Sena, G.; Presta, M. Zebrafish embryo as a tool to study tumor/endothelial cell cross-talk. Biochim. Biophys. Acta 2013, 1832, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, M.J.; Magner, J.A.; Ksander, B.R.; Dubovy, S.R. Uveal Melanoma Cell Lines: Where do they come from? (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2016, 114, T5. [Google Scholar] [PubMed]
- Gilbert, J.A. BGJ398 for FGFR-altered advanced cholangiocarcinoma. Lancet Oncol. 2018, 19, e16. [Google Scholar] [CrossRef]
- Zheng, L.; Pan, J. The Anti-malarial Drug Artesunate Blocks Wnt/beta-catenin Pathway and Inhibits Growth, Migration and Invasion of Uveal Melanoma Cells. Curr. Cancer Drug Targets 2018, 18, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Zuidervaart, W.; Pavey, S.; van Nieuwpoort, F.A.; Packer, L.; Out, C.; Maat, W.; Jager, M.J.; Gruis, N.A.; Hayward, N.K. Expression of Wnt5a and its downstream effector beta-catenin in uveal melanoma. Melanoma Res. 2007, 17, 380–386. [Google Scholar] [CrossRef]
- Chang, S.H.; Worley, L.A.; Onken, M.D.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Stei, M.M.; Loeffler, K.U.; Holz, F.G.; Herwig, M.C. Animal Models of Uveal Melanoma: Methods, Applicability, and Limitations. Biomed. Res. Int. 2016, 2016, 4521807. [Google Scholar] [CrossRef]
- Rodrigues, P.F.; Matarazzo, S.; Maccarinelli, F.; Foglio, E.; Giacomini, A.; Silva Nunes, J.P.; Presta, M.; Dias, A.A.M.; Ronca, R. Long Pentraxin 3-Mediated Fibroblast Growth Factor Trapping Impairs Fibrosarcoma Growth. Front. Oncol. 2018, 8, 472. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef]
- Helgadottir, H.; Hoiom, V. The genetics of uveal melanoma: current insights. Appl. Clin. Genet. 2016, 9, 147–155. [Google Scholar] [PubMed]
- Krejci, P.; Aklian, A.; Kaucka, M.; Sevcikova, E.; Prochazkova, J.; Masek, J.K.; Mikolka, P.; Pospisilova, T.; Spoustova, T.; Weis, M.; et al. Receptor tyrosine kinases activate canonical WNT/beta-catenin signaling via MAP kinase/LRP6 pathway and direct beta-catenin phosphorylation. PLOS One 2012, 7, e35826. [Google Scholar] [CrossRef] [PubMed]
- Buchtova, M.; Oralova, V.; Aklian, A.; Masek, J.; Vesela, I.; Ouyang, Z.; Obadalova, T.; Konecna, Z.; Spoustova, T.; Pospisilova, T.; et al. Fibroblast growth factor and canonical WNT/beta-catenin signaling cooperate in suppression of chondrocyte differentiation in experimental models of FGFR signaling in cartilage. Biochim. Biophys. Acta 2015, 1852, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Chua, V.; Orloff, M.; Teh, J.L.; Sugase, T.; Liao, C.; Purwin, T.J.; Lam, B.Q.; Terai, M.; Ambrosini, G.; Carvajal, R.D.; et al. Stromal fibroblast growth factor 2 reduces the efficacy of bromodomain inhibitors in uveal melanoma. EMBO Mol. Med. 2019, 11, e9081. [Google Scholar] [CrossRef] [PubMed]
- De Waard-Siebinga, I.; Blom, D.J.; Griffioen, M.; Schrier, P.I.; Hoogendoorn, E.; Beverstock, G.; Danen, E.H.; Jager, M.J. Establishment and characterization of an uveal-melanoma cell line. Int. J. Cancer 1995, 62, 155–161. [Google Scholar] [CrossRef]
- Verbik, D.J.; Murray, T.G.; Tran, J.M.; Ksander, B.R. Melanomas that develop within the eye inhibit lymphocyte proliferation. Int. J. Cancer 1997, 73, 470–478. [Google Scholar] [CrossRef]
- Chen, P.W.; Murray, T.G.; Uno, T.; Salgaller, M.L.; Reddy, R.; Ksander, B.R. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin. Exp. Metastasis 1997, 15, 509–518. [Google Scholar] [CrossRef]
- Rezzola, S.; Corsini, M.; Chiodelli, P.; Cancarini, A.; Nawaz, I.M.; Coltrini, D.; Mitola, S.; Ronca, R.; Belleri, M.; Lista, L.; et al. Inflammation and N-formyl peptide receptors mediate the angiogenic activity of human vitreous humour in proliferative diabetic retinopathy. Diabetologia 2017, 60, 719–728. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mel285 | 92.1 | Mel270 | OMM2.3 | |
|---|---|---|---|---|
| GNAQ (exons 4–5) | WT | Q209L (626 A > T) | Q209P (626 A > C) | Q209P (626 A > C) |
| GNA11 (exons 4–5) | Q209L | WT | WT | WT |
| BAP1 | WT | WT | WT | WT |
| BAP1 | Yes, low | Yes | Yes | Yes |
| SF3B1 | WT | WT | WT | WT |
| EIF1AX | WT | c.17G/A | WT | WT |
| Chr3 | Disomy 3 Loss 3p26-pter | Disomy 3 | Disomy 3 Loss 3p24 Loss 3q21.2-3q24 | Disomy 3 Loss 3p24 Loss 3q21.2-3q24 |
| Chr6 | Disomy 6p | Gain 6p | Tetrasomy 6p | Tetrasomy 6p |
| Chr8 | Disomy 8p Tetrasomy 8q | Gain 8q | Disomy 8 Extra 8q | Disomy 8 Extra 8q |
| Gene | Forward | Reverse |
|---|---|---|
| Fgf2 | 5’-CCTTCCCACCAGGCCACTTAA-3’ | 5’-GGTCCGTTTTGGATCCGAGTTT-3’ |
| Fgfr1 | 5’-GCTGACTCTGGCCTCTACGCT-3’ | 5’-CAGGATCTGGACATACGGCAA-3’ |
| Fgfr2 | 5’-CTGCCTGGTGGAGAATGAAT-3’ | 5’-CGCTGTAAACCTTGCAGACA-3’ |
| Ffgr3 | 5’-CTGAAGCACGTGGAAGTGAA-3’ | 5’-CCTCAAAGGTGACATTGTGC-3’ |
| Fgfr4 | 5’-ACTGTCAAATTCCGCTGTCC-3’ | 5’-AGCGAATGCTACCCAGAGAG-3’ |
| PTX3 | 5’-CATCTCCTTGCGATTCTGTTTTG-3’ | 5’-CCCATTCCGAGTGCTCCTGA-3’ |
| Gapdh | 5’-GAAGGTCGGTGTGAACGGATT-3’ | 5’-TGACTGTGCCGTTGAATTTG-3’ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezzola, S.; Ronca, R.; Loda, A.; Nawaz, M.I.; Tobia, C.; Paganini, G.; Maccarinelli, F.; Giacomini, A.; Semeraro, F.; Mor, M.; et al. The Autocrine FGF/FGFR System in both Skin and Uveal Melanoma: FGF Trapping as a Possible Therapeutic Approach. Cancers 2019, 11, 1305. https://doi.org/10.3390/cancers11091305
Rezzola S, Ronca R, Loda A, Nawaz MI, Tobia C, Paganini G, Maccarinelli F, Giacomini A, Semeraro F, Mor M, et al. The Autocrine FGF/FGFR System in both Skin and Uveal Melanoma: FGF Trapping as a Possible Therapeutic Approach. Cancers. 2019; 11(9):1305. https://doi.org/10.3390/cancers11091305
Chicago/Turabian StyleRezzola, Sara, Roberto Ronca, Alessandra Loda, Mohd Imtiaz Nawaz, Chiara Tobia, Giuseppe Paganini, Federica Maccarinelli, Arianna Giacomini, Francesco Semeraro, Marco Mor, and et al. 2019. "The Autocrine FGF/FGFR System in both Skin and Uveal Melanoma: FGF Trapping as a Possible Therapeutic Approach" Cancers 11, no. 9: 1305. https://doi.org/10.3390/cancers11091305
APA StyleRezzola, S., Ronca, R., Loda, A., Nawaz, M. I., Tobia, C., Paganini, G., Maccarinelli, F., Giacomini, A., Semeraro, F., Mor, M., & Presta, M. (2019). The Autocrine FGF/FGFR System in both Skin and Uveal Melanoma: FGF Trapping as a Possible Therapeutic Approach. Cancers, 11(9), 1305. https://doi.org/10.3390/cancers11091305