1. Introduction
BRAF is a member of the rapidly accelerated fibrosarcoma (RAF) kinase family, which transduces signals downstream of RAS via the mitogen-activated protein kinase (MAPK) pathway and induces cell growth and survival [
1]. Signals derived from activated receptor tyrosine kinases (RTKs) control the activation of RAS proteins through son-of-sevenless (SOS) and neurofibromin 1 (NF1), which activate and inactivate RAS, respectively. RAS can consequently stimulate RAF kinases through hetero- or homodimer formation, which in turn leads to MEK and ERK phosphorylation and activation. Under physiologic conditions, the MAPK pathway is tightly regulated through negative feedback loops. However, mutations in
BRAF can independently lead to uncontrolled cellular proliferation and cell survival through ERK signaling, and have been detected in melanoma (50–60%), colorectal cancer (10%), thyroid cancer (30–50%), serous ovarian cancer (30%), and non-small-cell lung cancer (NSCLC; 3%), amongst others [
2,
3]. Although these tumor types are located in different organs, it could be hypothesized that the functional consequence, and therefore the effect of treatment, of these mutations are similar.
In NSCLC,
BRAF V600 mutations have been found in 1–2% of patients, and are categorized as class I
BRAF mutations [
4]. Dual MAPK pathway inhibition using dabrafenib (BRAF inhibitor) plus trametinib (MEK inhibitor) achieved a 64% response rate and a median progression-free survival (PFS) of 10.9 months in
BRAF V600 mutation-positive NSCLC [
5], and the combined treatment was approved by the Food and Drug Administration (FDA) in 2017 [
6]. Effectivity of combined MEK and BRAF inhibition can be explained by the functional effect of class I
BRAF mutations.
BRAF V600D/E/K/R mutations result in a strong activation of the BRAF kinase and the MAPK pathway, while being RAS-independent. In fact, RAS activation is even suppressed in class I
BRAF mutations, due to a negative feedback loop after ERK activation [
1].
Next-generation sequencing (NGS) and other technical improvements have revealed that 50%-80% of
BRAF alterations in lung cancer are non-V600 and have considerably different signaling properties [
1,
7]. Non-V600
BRAF mutations can be subdivided into class II, with intermediate to high kinase activity and RAS independence, class III, with a lack of or impaired kinase activity, and “other”
BRAF mutations that have not been classified [
1,
4,
8,
9]. In addition, class III mutations harbor
RAS activating mutations,
NF1 tumor suppressor deletions, or depend on upstream receptor tyrosine kinase (RTK) signaling for cell growth [
7]. This dependence suggests that class III
BRAF mutant tumors are sensitive to RAS inhibition using RTK inhibitors [
10]. However, so far there are no effective targeted treatments available for patients harboring
KRAS,
NF1, and non-V600
BRAF alterations.
The non-receptor protein tyrosine phosphatase and scaffold protein SHP2 (
PTPN11) functions downstream of multiple RTKs, and promotes RAS activation after induction by growth factor signals [
7].
SHP2 mutations can consequently drive RAS/MAPK signaling, independent of RTK activation. Inhibiting SHP2 activity has been shown to suppress tumor cell growth by decreasing RAS/MAPK signaling, specifically in RTK-dependent cells [
11]. Since SHP2 comprises a convergent point in between RTK and downstream signaling, it has potential to be used as a new therapeutic target for cancer therapy. Different SHP2 inhibitors have been discovered and tested. SHP099 is a selective small-molecule inhibitor and stabilizes SHP2 through allosteric inhibition. In vitro and in vivo models have shown that SHP099 inhibits RAS/MAPK signaling in RTK driven cancer cells and suppresses cell proliferation. RMC-4550 is a more potent and selective allosteric inhibitor of SHP2, and was initially developed to treat esophageal tumors with
EGFR driver mutations [
12].
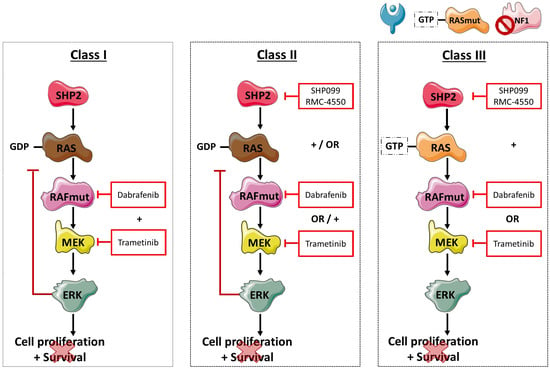
Based on the signaling mechanisms of the different
BRAF mutant classes, only class III
BRAF mutations are expected to be sensitive to single SHP2 inhibition due to the dependence of this specific
BRAF mutation on upstream signaling. Moreover,
KRAS G12C mutations and
NF1 loss-of-function mutations confer sensitivity to SHP2 inhibition [
7]. Bearing in mind that one in five NSCLC patients harbor one of these driver mutations, combining SHP2 inhibitors with BRAF or MEK inhibitors could have a huge impact on clinical outcome. Since class II
BRAF mutations are not RAS-dependent, it is unlikely that targeting SHP2 will affect cell proliferation. However, combining SHP2 with MAPK pathway inhibitors may induce synthetic lethality in this class of
BRAF mutations.
In the Spanish Lung Liquid versus Invasive Biopsy Program (SLLIP) study, genomic profiling was performed on plasma samples of 185 treatment-naïve advanced lung adenocarcinoma patients, using a 73-gene cell-free DNA (cfDNA) assay (Guardant360) (NCT03248089). A secondary aim of the study was the discovery of additional drivers and actionable mutations in plasma, particularly in the absence of
EGFR,
KRAS,
ALK, and
ROS1 alterations [
13].
In this study, we report on the frequency of different BRAF mutation classes that were detected in the cfDNA of patients from the SLLIP study and correlate the mutation classes with clinical outcome. In addition, we use in vitro studies in three distinct BRAF-mutant cell lines of different origin to highlight the relevance and importance of specific targeted treatment based on BRAF mutation class.
3. Discussion
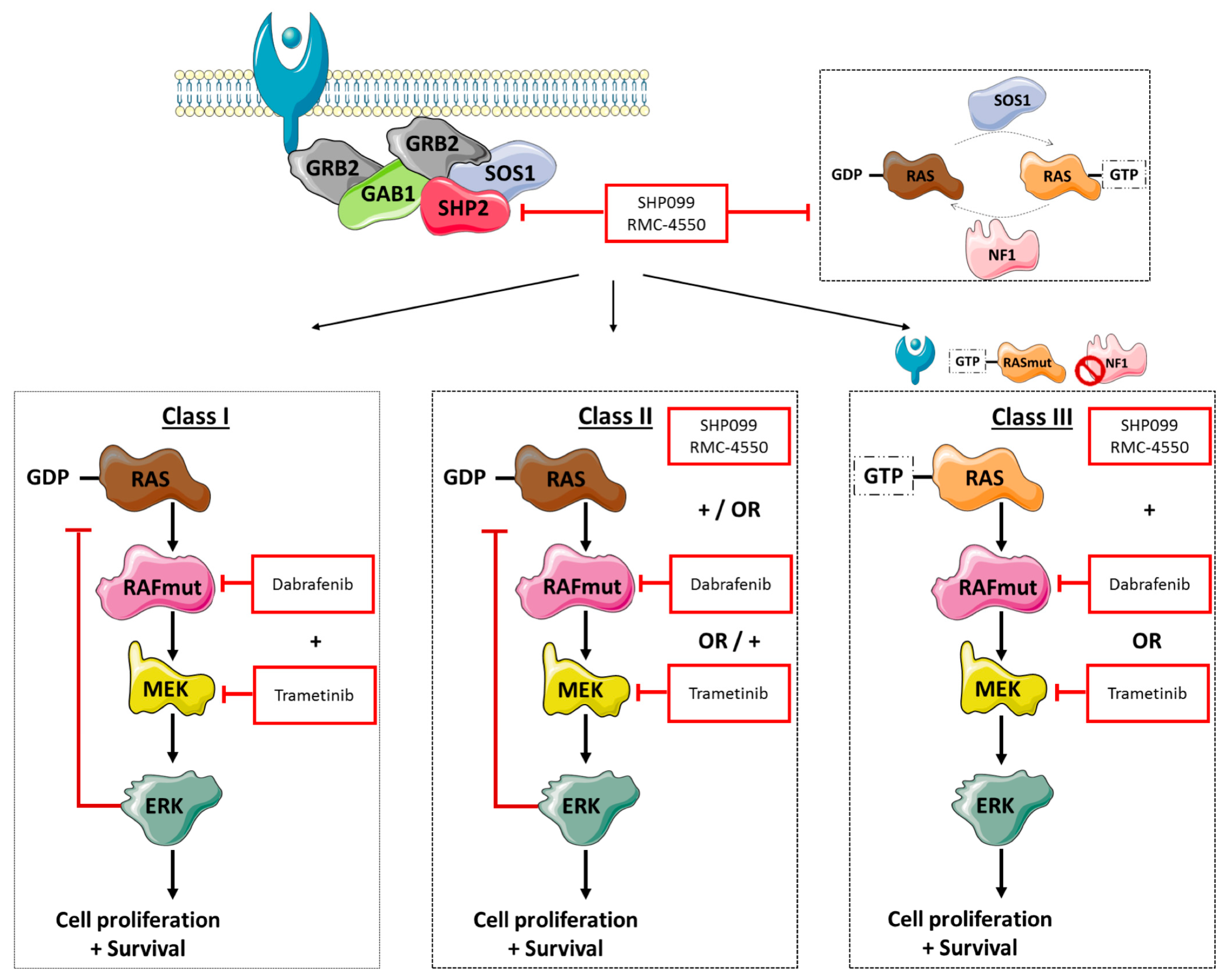
Under physiological conditions, RTK—and consequently GRB, GAB1, and SHP2 signaling—control the activation of RAS proteins through SOS1 and NF1. Once activated, RAS can then lead to downstream signaling through RAF dimer formation and MEK and ERK activation. Although the MAPK pathway is tightly regulated, mutations in
BRAF—with or without other co-alterations—can cause uncontrolled cellular proliferation and cell survival, as shown in
Figure 5. At present, NSCLC patients harboring V600
BRAF—or class I—mutations can be effectively treated with a combination of MEK and BRAF inhibitors. However, there are currently no approved targeted treatments for the 50–80% of patients with non-V600
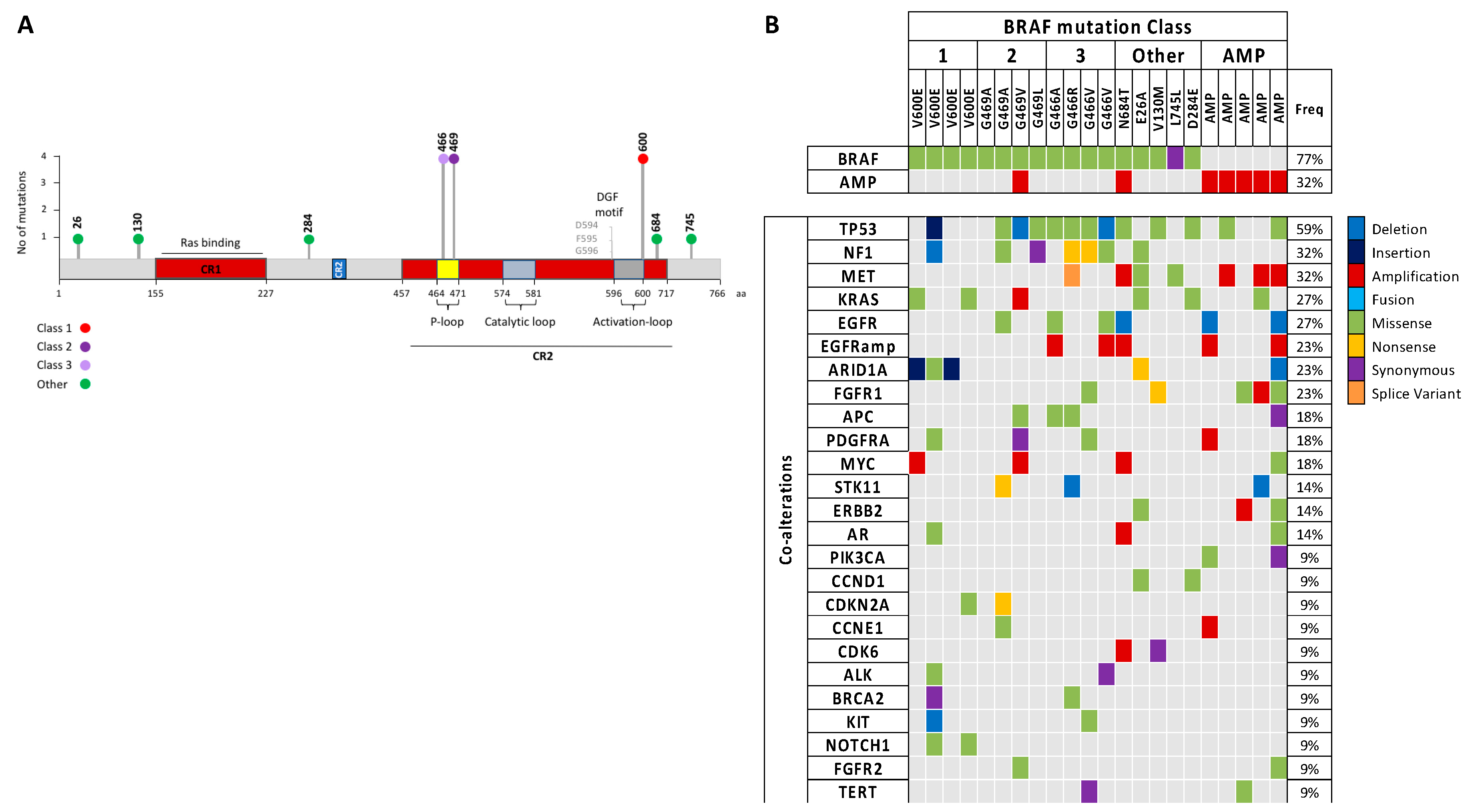
BRAF mutations. In this study, we confirm that complete exon sequencing with a cfDNA NGS panel enabled non-invasive detection of class I, II, III, and “other”
BRAF mutations plus amplifications. Primary analysis of the SLLIP study focused on comparing standard-of-care tissue genomic testing and cfDNA NGS testing for the detection of guideline-recommended biomarkers. Of note, none of the four
BRAF V600E mutations detected in plasma were identified via standard-of-care tissue genomic testing in the SLLIP study, highlighting the importance of liquid biopsies for mutation detection. In our patient cohort, approximately 80% of
BRAF altered patients comprised non-V600 alterations, including class II, class III,
BRAF amplifications, and “other” mutations that have not been classified yet. This preponderance of non-V600
BRAF alterations highlights the importance of developing new treatment options for patients harboring these mutations.
For effective use of targeted treatment, it is important to know which mutations are functionally relevant drivers of the tumor, and which mutations are passengers. Previous findings that class II
BRAF mutations are oncogenic drivers of lung adenocarcinoma [
14] are confirmed in our study, as the Guardant360 test sequences all 20 mutually exclusive oncogenic drivers in lung adenocarcinoma, and in most patients mutations in other known drivers did not co-occur with class II
BRAF mutations. Of the four class III
BRAF mutations, two
EGFR mutations were co-occurring and only one of those
EGFR mutations was found to be a driver mutation. Therefore, our results support that the class II and class III
BRAF alterations are indeed driver mutations in lung adenocarcinoma. In the Class I
BRAF-mutant cohort, additional
KRAS mutations were found in two out of four patients. In one patient, the
BRAF V600E was determined to be the driver mutation, and in the other patient, an additional
HRAS mutation was found to be the driver. Other passenger mutations were highly heterogeneous among patients, and although the groups consist of small numbers of patients, we found a trend of higher
P53 alteration frequency in class II and III
BRAF-mutant groups. Moreover, we hereby confirm that in the clinical setting, class III
BRAF mutations coexist with
NF1 nonsense and missense mutations.
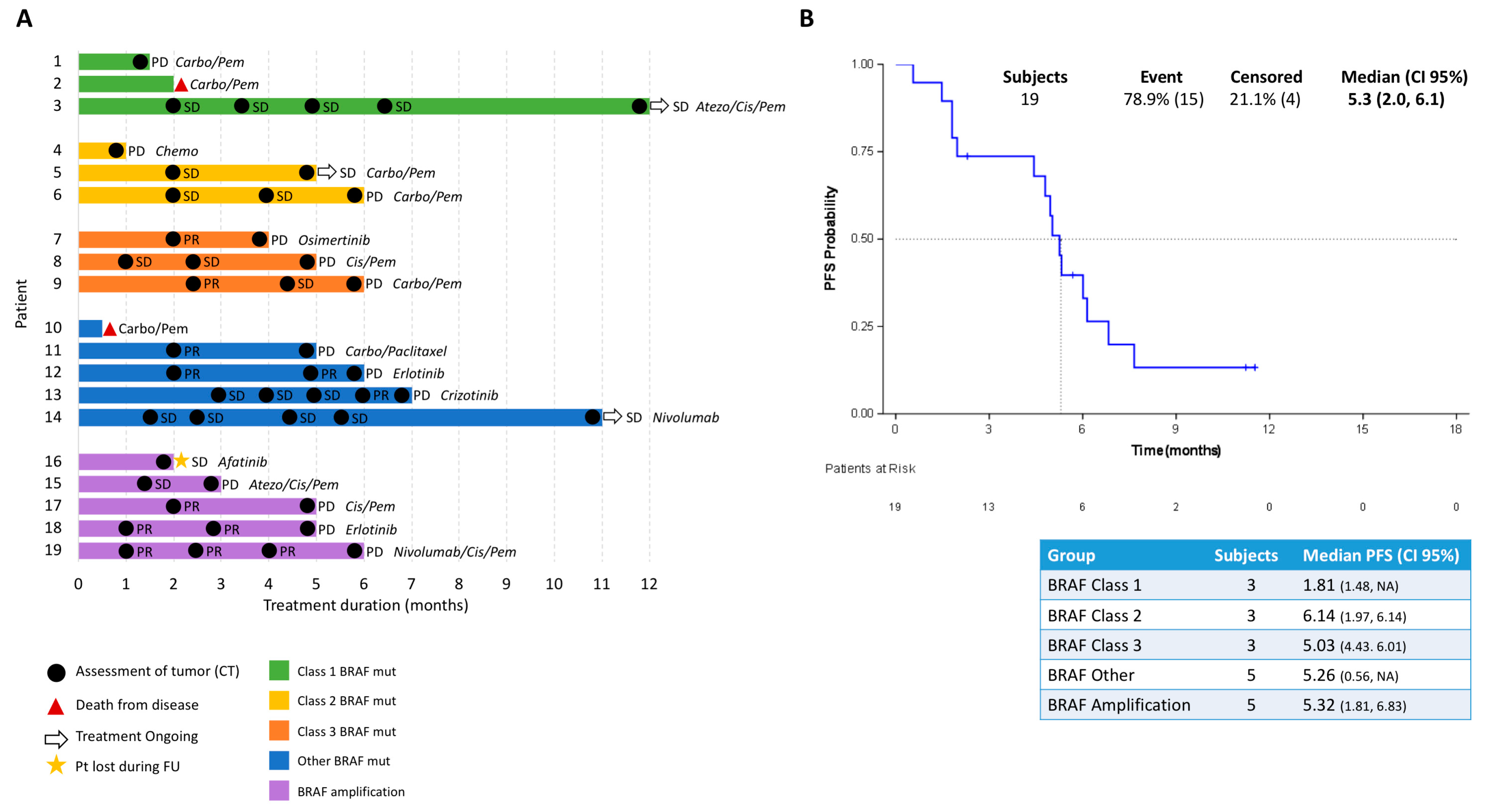
Previously published results indicate that non-V600
BRAF-mutant patients tend to have improved PFS to platinum-based chemotherapy, compared to
BRAF V600 mutation-positive patients [
15]. In contrast, another study reported shorter PFS for class II and III BRAF-mutant patients on chemotherapy, compared to class I
BRAF-mutant patients [
16]. Although our small cohorts cannot be used to draw any conclusions, non-V600 BRAF-mutant patients in the SLLIP study tend to have improved PFS to platinum-based chemotherapy. In addition, we found that patients treated with immunotherapy—independent of
BRAF mutation class—tend to have longer PFS than patients treated with targeted- or chemotherapy. Intriguingly, a recent study reported that MAPK alterations, including
BRAF and
SHP2 mutations, were enriched in patients that responded to the PD-1 inhibitors nivolumab and pembrolizumab [
17]. Since our patient cohort was small, further exploratory studies should be performed to validate these findings.
It is now known that
BRAF-mutation positive cancer is a heterogeneous disease, in which the location of the primary tumor may be less important than initially thought, and we hypothesize that focusing on specific alterations may be more important. Understanding the characteristics and divergent signaling properties of these different classes of mutations is critical for the development of effective therapeutic approaches. To this end, we explored the effect of dual treatment with RAF and MEK inhibitors on V600 and non-V600
BRAF-mutant cells, as shown in
Figure 5. While in patients dual treatment yielded a 64% response rate and a median PFS of 10.9 months in
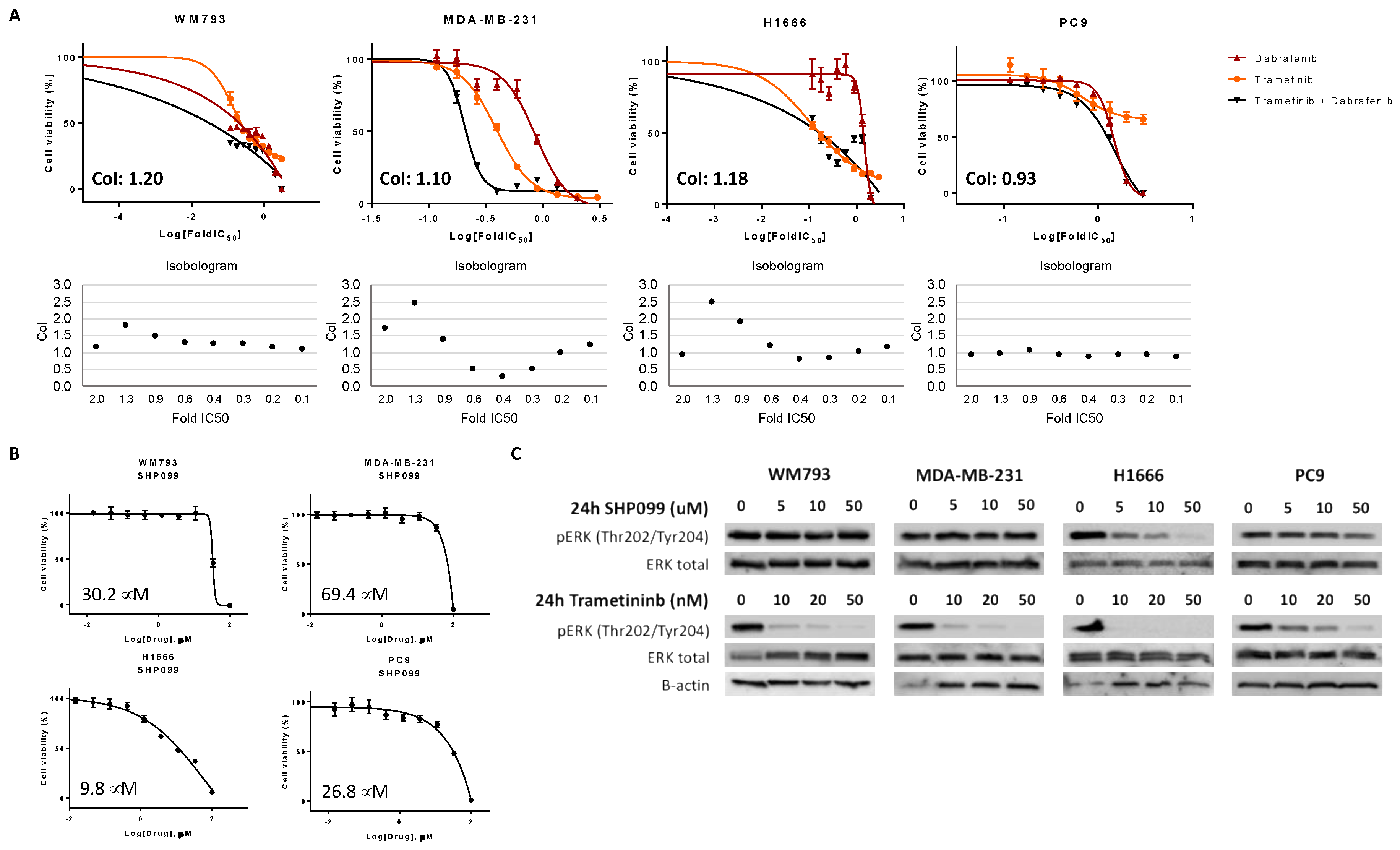
BRAF V600 mutation-positive NSCLC, combined RAF/MEK treatment in the class I
BRAF-mutant melanoma cell line (WM793) displayed additive to antagonistic effects. Class II
BRAF-mutant (MDA-MB-231) cells were found to be sensitive to dual RAF/MEK treatment, only at median drug concentrations. In addition, class I and class II
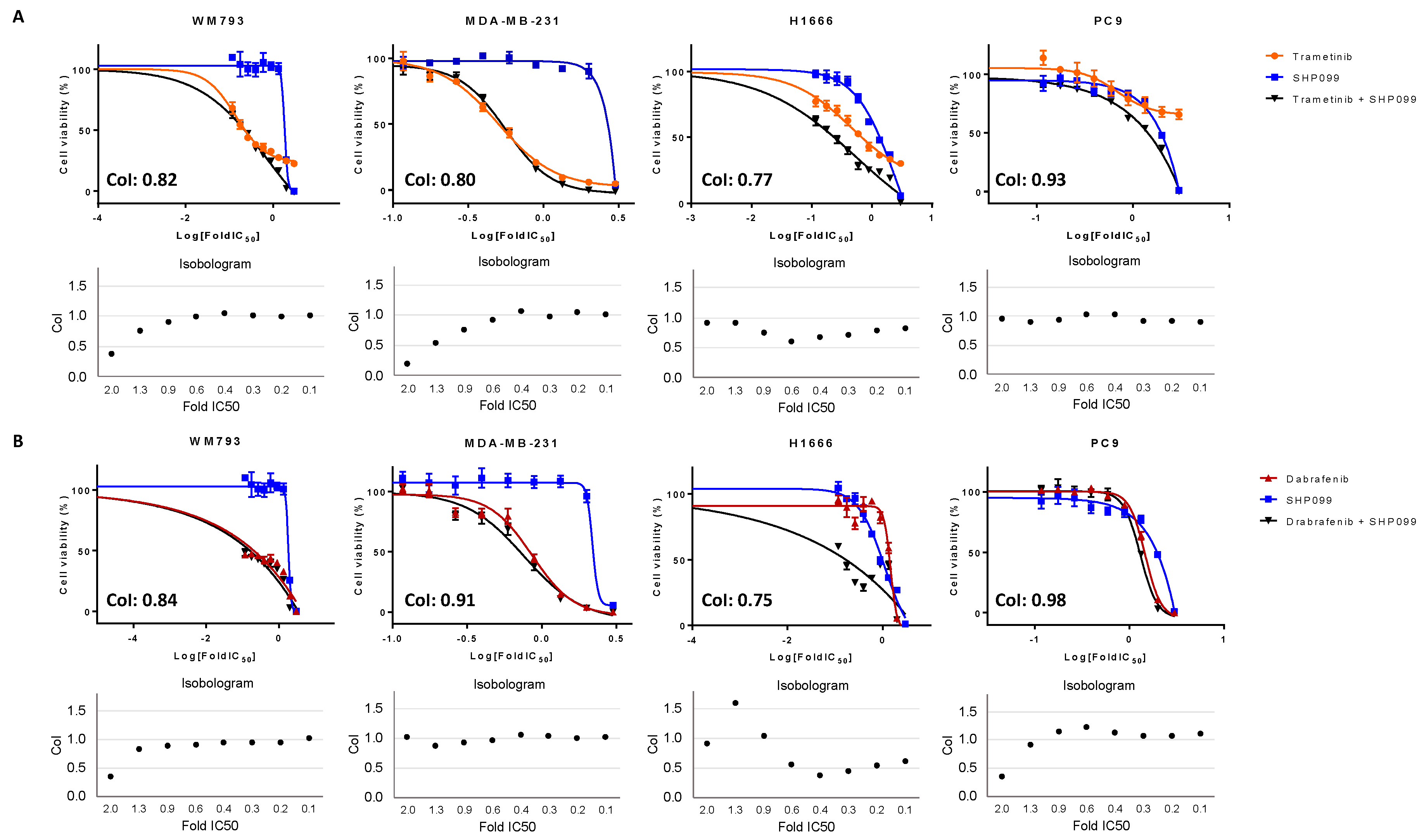
BRAF-mutant cells were intermediately sensitive and resistant, respectively, to single SHP2 inhibition, which can be explained by their RAS signaling independence [
7]. When RAF/MEK inhibitors were combined with SHP2 inhibition in the class I and II
BRAF-mutant and
BRAF WT cell lines, we obtained moderate synergistic effects. However, the main effect was derived from single RAF/MEK inhibition or single SHP2 inhibition. In the class III
BRAF-mutant cell line (H1666), we obtained antagonistic effects after combined RAF and MEK treatment. However, this cell line was sensitive to single SHP2 inhibition, due to the dependence on upstream RAS signaling. Cell viability experiments of combined RAF or MEK plus SHP2 inhibition in the class III
BRAF-mutant cell line indicated clear synergistic effects, revealing a possible new treatment approach for this
BRAF-mutant subtype. These results should be further exploited in vitro and in vivo, and if confirmed would warrant clinical development of class-specific therapy in
BRAF-altered patients.
4. Materials and Methods
4.1. Patients
This study provides a secondary analysis of the Spanish Lung Liquid vs. Invasive Biopsy Program (SLLIP) study, a multi-center prospective study (
clinicaltrials.gov registration NCT03248089) designed to evaluate the Guardant360® NGS assay in cfDNA compared to standard-of-care tissue-based genomic testing for the detection of guideline-recommended biomarkers in previously untreated NSCLC patients. The study was approved by the Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) and by the Ethics Committee of Investigación del Grupo Hospitalario Quirón (MedOPP125). Subjects were >18 years of age with a confirmed diagnosis of metastatic non-squamous NSCLC with no prior treatment for advanced NSCLC and an Eastern Collaborative Oncology Group (ECOG) performance status of 0–2. For this study, only patients that harbored BRAF alterations in baseline cfDNA analysis were included.
4.2. Procedures
Peripheral blood (20 mL) was collected in Streck® Cell-free DNA Blood Collection Tubes prior to treatment and sent to Guardant Health, Inc. Over the course of the study, plasma samples were collected and analyzed at two weeks after starting first-line therapy, and during progression or at the end of study (EOS) using the Guardant360 assay. The Guardant360 test employs digital sequencing to completely sequence the critical exons in 73 genes, including all 18 exons in BRAF, and identifies all four major classes of alterations: single nucleotide variants (SNVs), indels, and selected fusions and copy number amplifications. BRAF copy number amplification is reported as absolute plasma copy number and is a result of tissue copy number amplification and the degree of tumor DNA shed into the circulation. BRAF plasma copy number is filtered for focal amplifications through comparison to three other genes (EGFR, MET, CDK6) that reside, with BRAF, on chromosome 7. Clinical data, including overall response rate (ORR) and PFS were collected for all patients.
4.3. Data Analysis
Clinical data was used to explore the effect of BRAF class on PFS. The PFS in this study was defined as the time from the start of the first line treatment, to the date of progression or death. Patients who did not progress, neither die, were censored at the last visit date where their status was confirmed to be alive and without progressive disease (PD). Patients that did not start first-line treatment were excluded from analysis. Due to the small sample sizes, data analysis was considered to be exploratory.
4.4. Chemicals and Reagents
Human lung adenocarcinoma H1666 cells, harboring BRAF G466V (Class 3) mutation, human TNBC, MDA-MB-231 cells, harboring BRAF G464V (Class 2), and human melanoma WM793 cells, harboring BRAF V600E, were purchased from the American Type Culture Collection (ATCC). Human lung adenocarcinoma PC9 cells, harboring EGFR exon 19 deletion, but no BRAF mutations were provided by Mayumi Ono, (Kyushu University, Fukuoka, Japan). All cell lines were maintained in RPMI (Roswell Park Memorial Institute medium) 1640 supplemented with 1% Gibco penicillin/streptomycin/glutamine (Thermo Fisher Scientific, Waltham, MA, USA) and 10% Gibco fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) in a 5% CO2 37 °C cell culture incubator and routinely evaluated for mycoplasma contamination. The MEK inhibitor trametinib, BRAF inhibitor dabrafenib, and both SHP2 inhibitors SHP099 and RMC-4550 were bought from Selleck Chemicals (Houston, TX, USA). Drugs were prepared in dimethylsulfoxide (DMSO) at a concentration of 10–100 mM stock solutions and stored at −20 °C. Further dilutions were made in culture medium to final concentration before use. Antibodies rabbit anti-ERK (#9102) and rabbit anti-phospho-ERK (T202/Y204; #9101) were purchased from Cell Signaling (Leiden, The Netherlands) and diluted 1:1000. Mouse anti-Beta-actin (#A5441) was purchased from Sigma Aldrich/Merck (Darmstadt, Germany) and diluted 1:5000. Secondary horseradish peroxidase (HRP)-linked goat anti-rabbit (from donkey, NA934-1ML) and HRP-linked goat anti-mouse (from sheep; NA931-1ML) were purchased from GE Healthcare Life Sciences (Amersham, UK) and diluted 1:5000.
4.5. Cell Viability Assay
Cells were seeded in 96-well plates at the following densities: 1.0 × 103 (PC9), 1.5 × 103 (WM793), 2.5 × 103 (H1666), and 1.5 × 103 (MDA-MB-231), and incubated for 24 h. Cell viability was assessed using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma Aldrich, St. Louis, MO, USA), after treatment with serial drug dilutions. For the half maximal inhibitory concentration (IC50) determination, MTT viability assays were performed using SHP099 (all cell lines) and RMC-4550 (H1666 and MDA-MB-231) doses ranging from 0–100 μM and trametinib doses ranging from 0–2 μM. For dabrafenib, doses ranged from 0–500 μM for MDA-MB-231, PC9 and WM793 and 0–600 μM for H1666.
To determine the combined effect of trametinib and dabrafenib on cell viability, cells were treated with trametinib doses ranging from 0 to 2 μM, and dabrafenib doses ranging from 0 to 500 μM (MDA-MB-231, PC9, and WM793) or 0–600 μM (H1666), or with the combination of both. In addition, cells were treated with trametinib doses ranging from 0 to 2 μM, and SHP099 or RMC-4550 doses ranging from 0 to 100 μM, or with a combination of trametinib plus SHP099 or RMC-4550. Finally, cell lines were treated with dabrafenib doses from 0 to 500 μM (MDA-MB-231, PC9, and WM793) or 0–600 μM (H1666), and SHP099 or RMC-4550 doses ranging from 0 to 100 μM, or with a combination of dabrafenib plus SHP099 or RMC-4550. After 72 h of treatment incubation, 0.5 mg/ml of MTT was added to the medium in the wells for 2 h at 37 °C and formazan crystals in viable cells were solubilized with 100 μl DMSO and spectrophotometrically quantified using a microplate reader (Varioskan Flash; Thermo Fisher Scientific, Waltham, MA, USA) at 565 nm of absorbance. Fractional survival was then calculated as a percentage to control cells. Data of combined drug effects were subsequently analyzed by the Chou and Talalay method [
18,
19]. Combination Index (CoI) values <1, =1, and >1 indicated synergism, additive effect, and antagonism, respectively. All experiments were performed in biological triplicates.
4.6. Western Blotting
For immunoblotting experiments, cells were seeded in T25 flasks (Sarstedt, Newton, NC, USA). Cell amounts were 0.5 million per flask (PC9, MDA-MB-231, and H1666), 0.75 million per flask (WM793), after 24 h, cells were either treated with increasing doses of SHP099 (5, 10, and 50 μM), or with increasing doses of trametinib (10, 20, and 50 nM). Untreated cells received an equivalent dose of vehicle (DMSO). After 24 h, cells were washed with cold PBS and re-suspended in ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM tris-hydrochloric acid in pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 150 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid, 1 mM sodium vanadate, and 50 mM sodium fluoride) containing protease inhibitor mixture (Roche Applied Science, Penzberg, Germany). Following cell lysis by sonication and centrifugation at 18,620× g for 10 min at 4 °C, the resulting supernatant was collected as the total cell lysate. Briefly, the lysates containing 30 μg proteins were electrophoresed on 10% SDS-polyacrylamide gels (Life Technologies, Carlsbad, CA, USA) and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories Inc., Hercules, CA, USA). Membranes were blocked in Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA). All target proteins were immunoblotted with appropriate primary and horseradish peroxidase (HRP)-conjugated secondary antibodies. Chemiluminescent (HRP-conjugated) bands were detected in a ChemiDoc MP Imaging System (Bio-Rad Laboratories Inc.). β-actin was used as an internal control to confirm equal gel loading. Experiments were performed in biological triplicates with similar results, and representative blots were shown.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}