ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line

Abstract
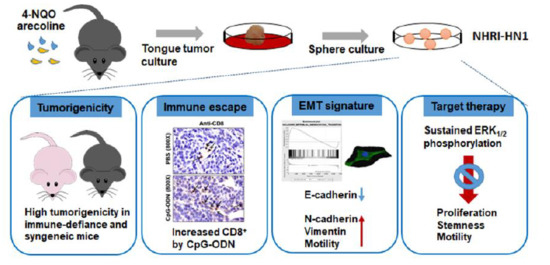
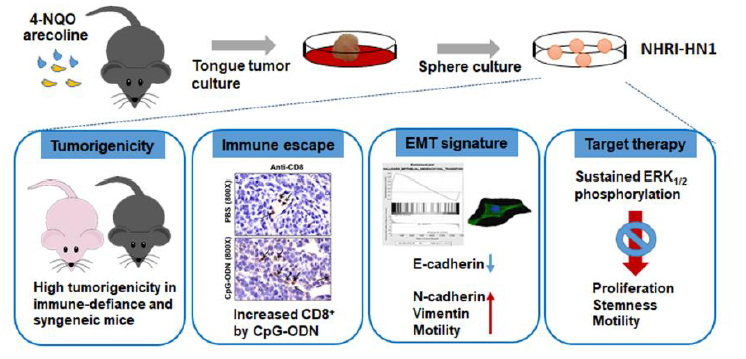
:
1. Introduction
2. Results
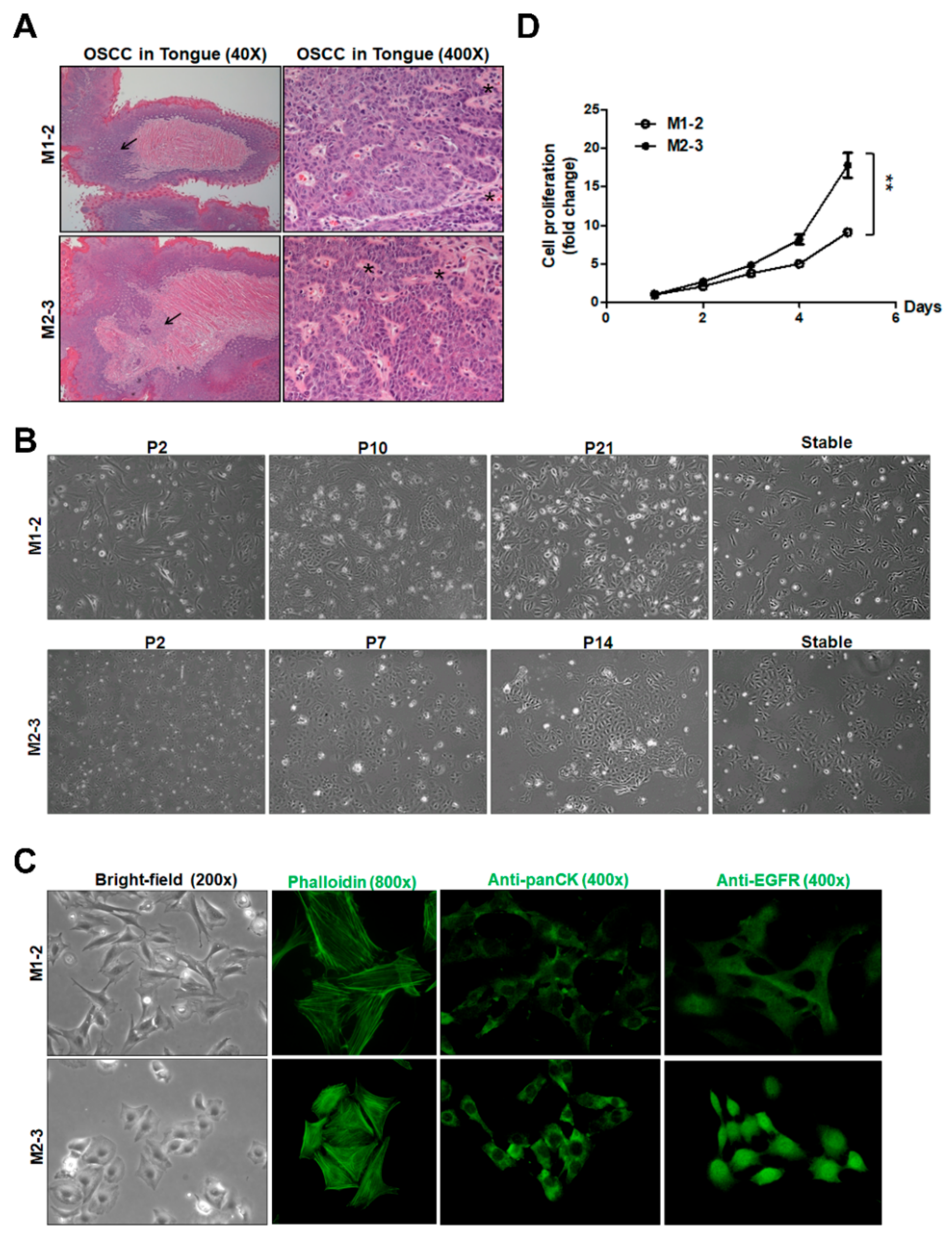
2.1. Establishment of Two Mouse OSCC Cell Lines from Carcinogen-Induced OSCC Mice
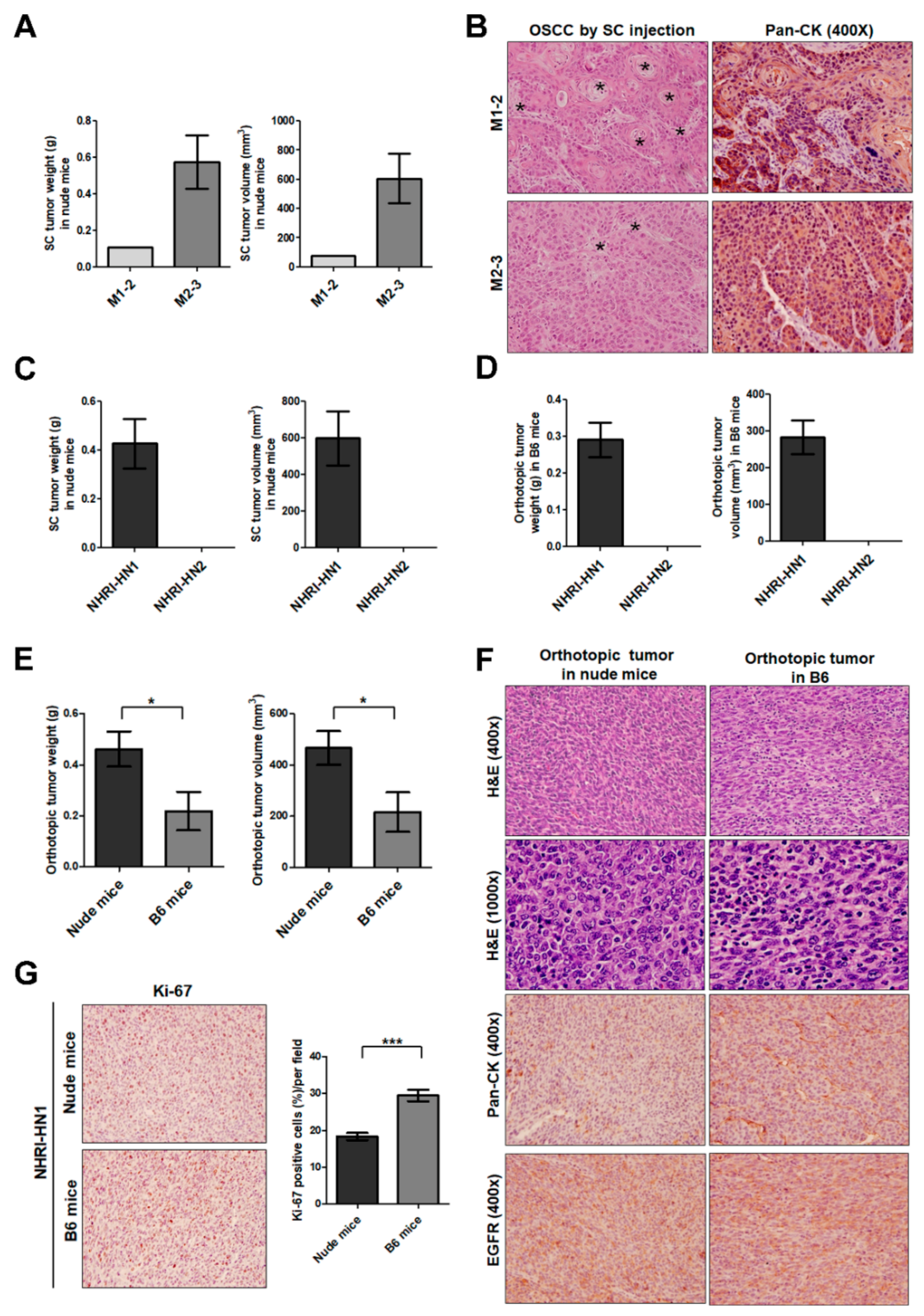
2.2. Tumor Growth of Mouse OSCC Cells in Immunocompromised Mice and Syngeneic Mice
2.3. Tumor Growth of In Vitro Selected Mouse OSCC Cells in Immunocompromised and Immunocompetent Mice
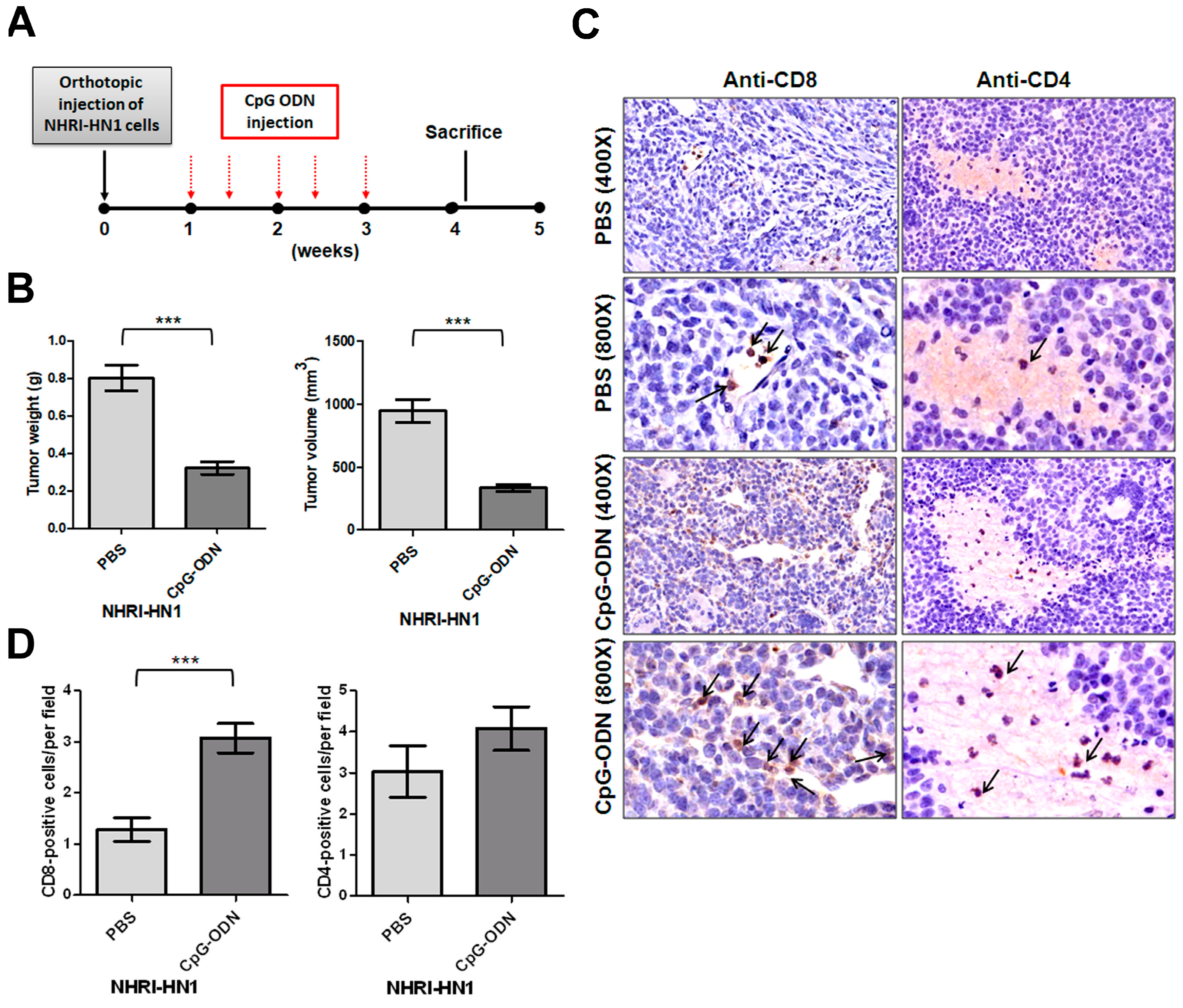
2.4. Effects of Immune Modulation on Syngeneic OSCC Tumors
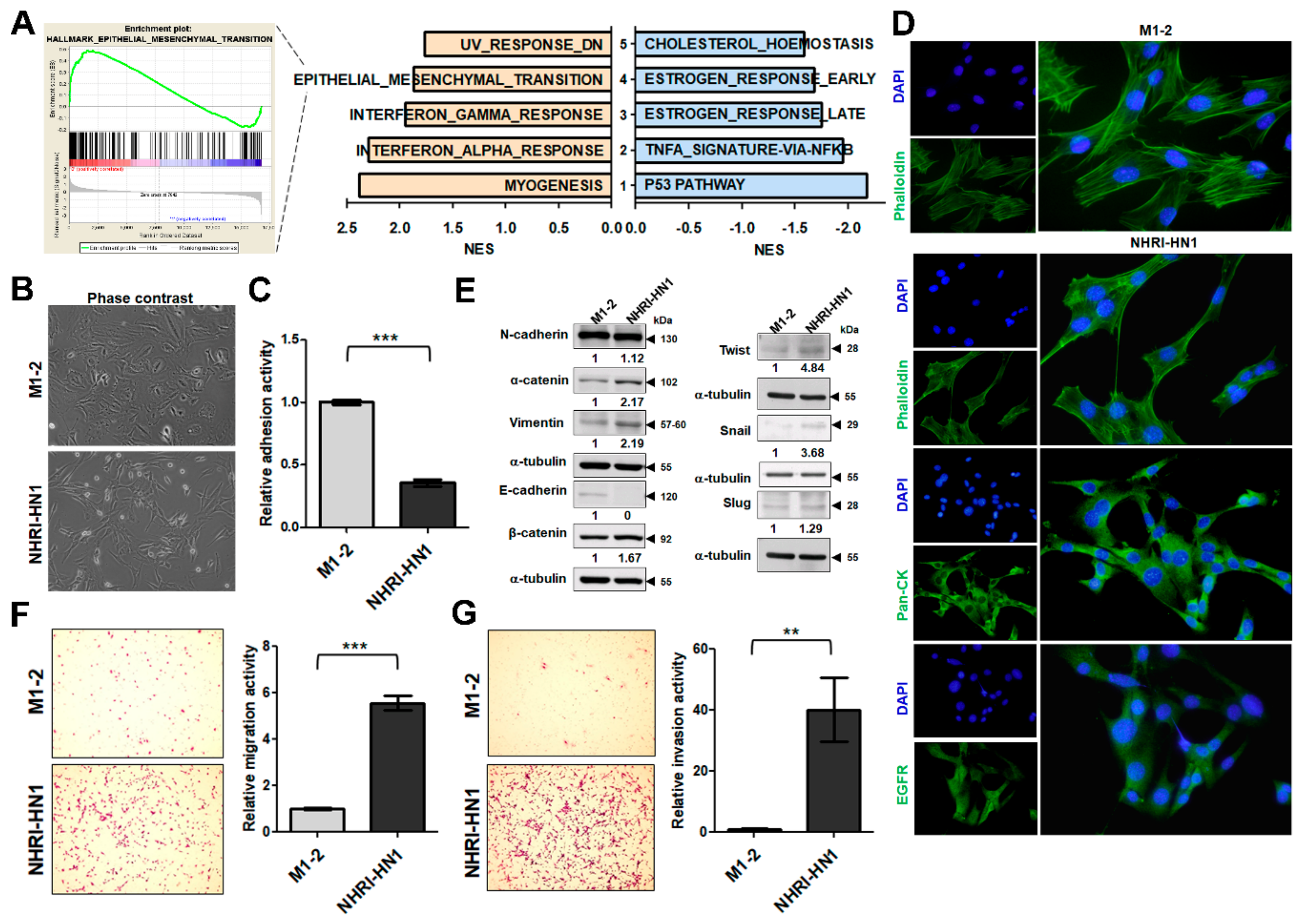
2.5. Epithelial Mesenchymal Transition, Migration and Invasion in NHRI-HN1 Cells
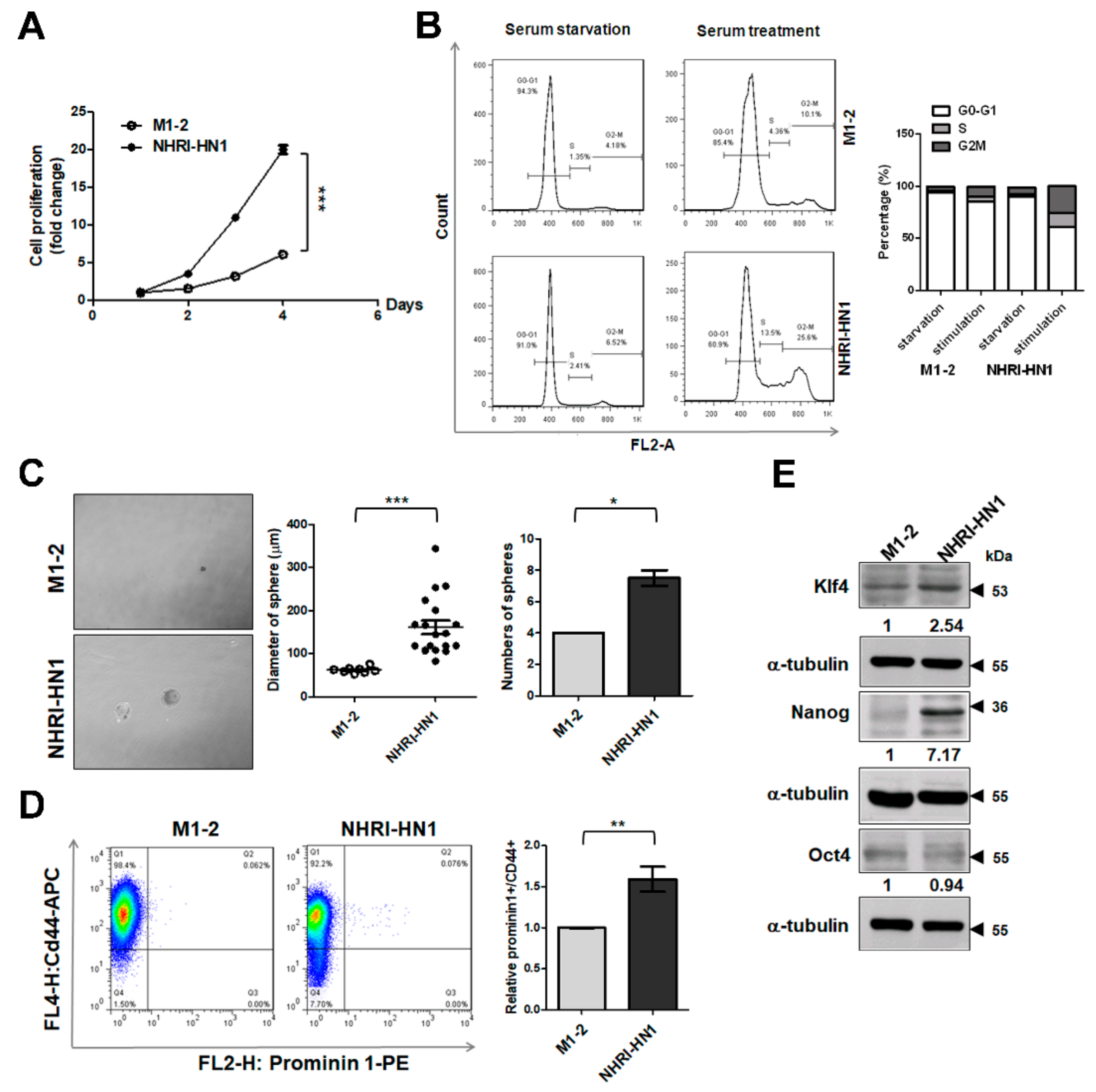
2.6. Cell Growth and Cancer Stemness Characteristics of NHRI-HN1 Cells
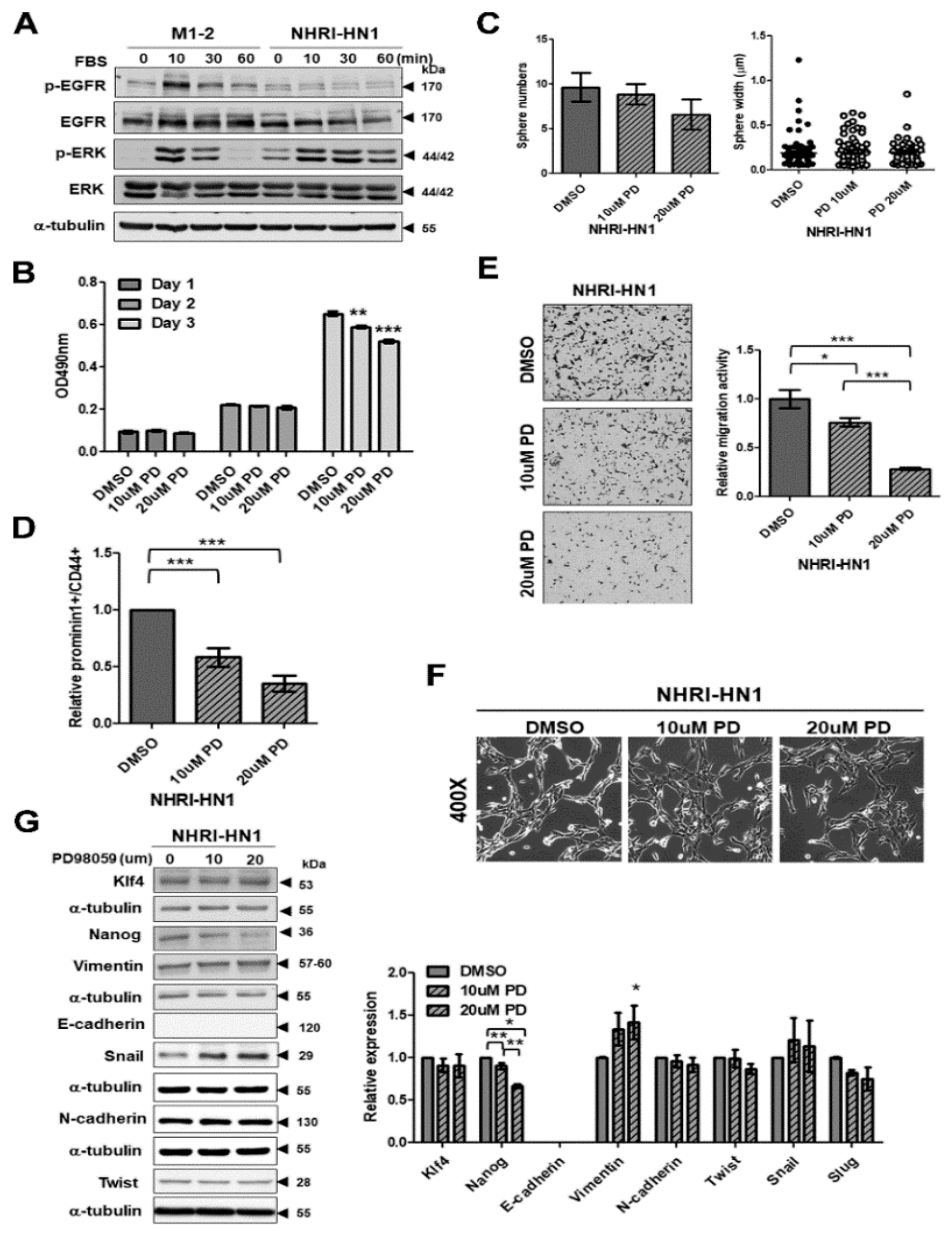
2.7. Inhibition of ERK Activation Impairs Cancer Stemness and Migration in NHRI-HN1 Cells
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mouse Tongue Squamous Cell Carcinoma
4.3. In Vitro Selection and Cell Culture
4.4. DNA Extraction and Amplification of Cox I and Short Tandem Repeat Sequences With Polymerase Chain Reaction
4.5. Immunohistochemistry
4.6. Morphological Analysis
4.7. Immunofluorescence
4.8. Cell Proliferation
4.9. Subcutaneous Tumor Growth In Nude Mice
4.10. Orthotopic Injection in Syngeneic Mice
4.11. Orthotopic Injection of NHRI-HN1 Cells in Nude Mice and Syngeneic Mice
4.12. CpG Oligodeoxynucleotide (CpG-ODN) Treatment
4.13. Gene Expression and Pathway Analysis
4.14. Adhesion assay
4.15. Immunoblot Assay
4.16. Migration and Invasion Assays
4.17. Cell Cycle Analysis
4.18. Sphere Formation Assay
4.19. Detection of Cancer Stem Cells by Fluorescence Activated Cell Sorting (FACS) Analysis
4.20. EGFR Sequencing
4.21. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer. J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagan, J.V.; Scully, C. Recent advances in Oral Oncology 2007: Epidemiology, aetiopathogenesis, diagnosis and prognostication. Oral Oncol. 2008, 44, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Brunin, F.; Mosseri, V.; Jaulerry, C.; Point, D.; Cosset, J.M.; Rodriguez, J. Cancer of the base of the tongue: Past and future. Head Neck 1999, 21, 751–759. [Google Scholar] [CrossRef]
- Mehra, R.; Cohen, R.B.; Burtness, B.A. The role of cetuximab for the treatment of squamous cell carcinoma of the head and neck. Clin. Adv. Hematol. Oncol. 2008, 6, 742–750. [Google Scholar]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet. Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Ann. Rev. Pathol. 2010, 6, 95–119. [Google Scholar] [CrossRef]
- Dranoff, G. Experimental mouse tumour models: What can be learnt about human cancer immunology? Nat. Rev. Immunol. 2011, 12, 61–66. [Google Scholar] [CrossRef]
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2014, 517, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Sasahira, T.; Kirita, T. Hallmarks of Cancer-Related Newly Prognostic Factors of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Liu, C.J.; Lin, L.H.; Chou, C.H.; Yeh, L.Y.; Lin, S.C.; Chang, K.W. Establishing of mouse oral carcinoma cell lines derived from transgenic mice and their use as syngeneic tumorigenesis models. BMC Cancer 2019, 19, 281. [Google Scholar] [CrossRef] [PubMed]
- Malik, U.U.; Zarina, S.; Pennington, S.R. Oral squamous cell carcinoma: Key clinical questions, biomarker discovery, and the role of proteomics. Arch. Oral. Biol. 2016, 63, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; Lewis, J.S., Jr.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; Uppaluri, R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012, 72, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect. Biol. 2012, 4, a011908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Filho, M.S.; Nor, J.E. The biology of head and neck cancer stem cells. Oral Oncol. 2011, 48, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Quentmeier, H.; Osborn, M.; Reinhardt, J.; Zaborski, M.; Drexler, H.G. Immunocytochemical analysis of cell lines derived from solid tumors. J. Histochem. Cytochem. 2001, 49, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.C.; Zhang, S.; Ishii, G.; Endoh, Y.; Kodama, K.; Miyamoto, S.; Hayashi, R.; Ebihara, S.; Cho, J.S.; Ochiai, A. Predictive markers for late cervical metastasis in stage I and II invasive squamous cell carcinoma of the oral tongue. Clin. Cancer Res. 2004, 10, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Bhardwaj, N.; Kohrt, H.E.; Brody, J.D. In situ vaccination for the treatment of cancer. Immunotherapy 2016, 8, 315–330. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef]
- Chiou, S.H.; Yu, C.C.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Tsai, T.H.; Chou, S.H.; Chien, C.S.; Ku, H.H.; Lo, J.F. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosunen, A.; Pirinen, R.; Ropponen, K.; Pukkila, M.; Kellokoski, J.; Virtaniemi, J.; Sironen, R.; Juhola, M.; Kumpulainen, E.; Johansson, R.; et al. CD44 expression and its relationship with MMP-9, clinicopathological factors and survival in oral squamous cell carcinoma. Oral Oncol. 2007, 43, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Shah, K.A.; Shah, M.J.; Kothari, K.C.; Rawal, R.M. Cancer stem cells and stemness markers in oral squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2014, 15, 8549–8556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.W.; Pei, R.J.; Tseng, H.C.; Yeh, K.T.; Chan, H.C.; Lee, M.R.; Lin, C.; Hsieh, W.T.; Kao, M.C.; Tsai, M.H.; et al. Co-treating with arecoline and 4-nitroquinoline 1-oxide to establish a mouse model mimicking oral tumorigenesis. Chem. Biol. Interact. 2010, 183, 231–237. [Google Scholar] [CrossRef]
- Lee, C.H.; Chang, J.S.; Syu, S.H.; Wong, T.S.; Chan, J.Y.; Tang, Y.C.; Yang, Z.P.; Yang, W.C.; Chen, C.T.; Lu, S.C.; et al. IL-1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J. Cell Physiol. 2014, 230, 875–884. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, 185–198. [Google Scholar] [CrossRef]
- Baines, J.; Celis, E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin. Cancer Res. 2003, 9, 2693–2700. [Google Scholar]
- Yang, T.L.; Wu, C.T.; Ko, J.Y.; Wang, C.P.; Lou, P.J.; Chang, Y.L. Significance of tumor satellite variables in reflecting the epithelial-mesenchymal transition of tongue cancer. Oral Oncol. 2011, 47, 720–724. [Google Scholar] [CrossRef]
- Chaw, S.Y.; Abdul Majeed, A.; Dalley, A.J.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers-E-cadherin, beta-catenin, APC and Vimentin--in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Sheu, J.J.; Hua, C.H.; Wan, L.; Lin, Y.J.; Lai, M.T.; Tseng, H.C.; Jinawath, N.; Tsai, M.H.; Chang, N.W.; Lin, C.F.; et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009, 69, 2568–2576. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Shin, M.S.; Sakimura, A.; Zhou, Y.; Tanaka, T.; Kawanishi, M.; Kawasaki, Y.; Yokoyama, S.; Koizumi, K.; Saiki, I.; et al. Inverse correlation between Thr-669 and constitutive tyrosine phosphorylation in the asymmetric epidermal growth factor receptor dimer conformation. Cancer Sci. 2013, 104, 1315–1322. [Google Scholar] [CrossRef]
- Mishima, K.; Inoue, K.; Hayashi, Y. Overexpression of extracellular-signal regulated kinases on oral squamous cell carcinoma. Oral Oncol. 2002, 38, 468–474. [Google Scholar] [CrossRef]
- Fremin, C.; Meloche, S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Yen, Y.C.; Hsiao, J.R.; Jiang, S.S.; Chang, J.S.; Wang, S.H.; Shen, Y.Y.; Chen, C.H.; Chang, I.S.; Chang, J.Y.; Chen, Y.W. Insulin-like growth factor-independent insulin-like growth factor binding protein 3 promotes cell migration and lymph node metastasis of oral squamous cell carcinoma cells by requirement of integrin beta1. Oncotarget 2015, 6, 41837–41855. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhu, G.; Li, Y.; Padia, R.N.; Dong, Z.; Pan, Z.K.; Liu, K.; Huang, S. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009, 69, 9228–9235. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.H.; Kim, B.W.; Song, K.H.; Cho, H.; Lee, Y.H.; Kim, J.H.; Chung, J.Y.; Hewitt, S.M.; Seong, S.Y.; Mao, C.P.; et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J. Clin. Invest. 2012, 122, 4077–4093. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.H.; Lee, Y.H.; Jeon, J.H.; Kang, T.H.; Mao, C.P.; Wu, T.C.; Kim, T.W. Cancer vaccination drives Nanog-dependent evolution of tumor cells toward an immune-resistant and stem-like phenotype. Cancer Res. 2012, 72, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.E.; Foster, S.; Betts, D.; Marnock, W.E. DOK, a cell line established from human dysplastic oral mucosa, shows a partially transformed non-malignant phenotype. Int. J. Cancer 1992, 52, 896–902. [Google Scholar] [CrossRef]
- Lu, Y.C.; Chen, Y.J.; Wang, H.M.; Tsai, C.Y.; Chen, W.H.; Huang, Y.C.; Fan, K.H.; Tsai, C.N.; Huang, S.F.; Kang, C.J.; et al. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer Prev. Res. 2012, 5, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Liu, C.J.; Chiu, C.P.; Chang, S.M.; Lu, S.Y.; Chen, Y.J. Establishment of OC3 oral carcinoma cell line and identification of NF-kappa B activation responses to areca nut extract. J. Oral Pathol. Med. 2004, 33, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Lim, J.S.; Shyu, K.W.; Meng, C.L. Direct cytotoxicity of garlic on human oral cancer cells. Zhonghua Ya Yi Xue Hui Za Zhi 1988, 7, 13–18. [Google Scholar] [PubMed]
- Takahashi, K.; Kanazawa, H.; Akiyama, Y.; Tazaki, S.; Takahara, M.; Muto, T.; Tanzawa, H.; Sato, K.-I. Establishment and characterization of a cell line (SAS) from poorly differentiated human squamous cell carcinoma of the tongue. J. Jap. Stomatol. Soc. 1989, 38, 20–28. [Google Scholar]
- Yen, Y.C.; Shiah, S.G.; Chu, H.C.; Hsu, Y.M.; Hsiao, J.R.; Chang, J.Y.; Hung, W.C.; Liao, C.T.; Cheng, A.J.; Lu, Y.C.; et al. Reciprocal regulation of microRNA-99a and insulin-like growth factor I receptor signaling in oral squamous cell carcinoma cells. Mol. Cancer 2014, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Parodi, B.; Aresu, O.; Bini, D.; Lorenzini, R.; Schena, F.; Visconti, P.; Cesaro, M.; Ferrera, D.; Andreotti, V.; Ruzzon, T. Species identification and confirmation of human and animal cell lines: A PCR-based method. Biotechniques 2002, 32, 438–440. [Google Scholar] [CrossRef]
- Almeida, J.L.; Hill, C.R.; Cole, K.D. Mouse cell line authentication. Cytotechnology 2013, 66, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.S.; Chu, H.C.; Yen, Y.C.; Lewis, B.C.; Chen, Y.W. Kruppel-like factor 4, a tumor suppressor in hepatocellular carcinoma cells reverts epithelial mesenchymal transition by suppressing slug expression. PLoS ONE 2012, 7, e43593. [Google Scholar] [CrossRef]
- Chen, Y.W.; Paliwal, S.; Draheim, K.; Grossman, S.R.; Lewis, B.C. p19Arf inhibits the invasion of hepatocellular carcinoma cells by binding to C-terminal binding protein. Cancer Res. 2008, 68, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Klimstra, D.S.; Mongeau, M.E.; Tatem, J.L.; Boyartchuk, V.; Lewis, B.C. Loss of p53 and Ink4a/Arf cooperate in a cell autonomous fashion to induce metastasis of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 7589–7596. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Boyartchuk, V.; Lewis, B.C. Differential roles of insulin-like growth factor receptor- and insulin receptor-mediated signaling in the phenotypes of hepatocellular carcinoma cells. Neoplasia 2009, 11, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Yeh, D.W.; Lai, C.Y.; Liu, Y.L.; Lu, C.H.; Tseng, P.H.; Yuh, C.H.; Yu, G.Y.; Liu, S.J.; Leng, C.H.; Chuang, T.H. CpG-oligodeoxynucleotides developed for grouper toll-like receptor (TLR) 21s effectively activate mouse and human TLR9s mediated immune responses. Sci. Rep. 2017, 7, 17297. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Wu, W.L.; Jang, C.W.; Yen, Y.C.; Wang, S.H.; Tsai, F.Y.; Shen, Y.Y.; Chen, Y.W. Interferon-stimulated gene 15 modulates cell migration by interacting with Rac1 and contributes to lymph node metastasis of oral squamous cell carcinoma cells. Oncogene 2019, 38, 4480–4495. [Google Scholar] [CrossRef]
- Brown, L.D.; Cai, T.T.; DasGupta, A. Interval estimation for a binomial proportion. Statist. Sci. 2001, 16, 101–133. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Nude Mice (Subcutaneous Injection) | B6 Mice (Orthotopic Injection) | ||
|---|---|---|---|---|
| Tumors/No. of Mice | Tumorigenesis (%) | Tumors/No. of Mice | Tumorigenesis (%) | |
| M1-2 | 1/3 | 33.33 | 0/3 | 0 |
| M2-3 | 2/3 | 66.67 | 0/3 | 0 |
| NHRI-HN1 | 4/4 | 100 | 3/3 | 100 |
| NHRI-HN2 | 0/4 | 0 | 0/3 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-L.; Liu, K.-J.; Jang, C.-W.; Hsu, C.-C.; Yen, Y.-C.; Liu, Y.-L.; Chuang, T.-H.; Wang, S.-H.; Fu, Y.-K.; Kuo, C.-C.; et al. ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers 2020, 12, 61. https://doi.org/10.3390/cancers12010061
Chen Y-L, Liu K-J, Jang C-W, Hsu C-C, Yen Y-C, Liu Y-L, Chuang T-H, Wang S-H, Fu Y-K, Kuo C-C, et al. ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers. 2020; 12(1):61. https://doi.org/10.3390/cancers12010061
Chicago/Turabian StyleChen, Yu-Lin, Ko-Jiunn Liu, Chuan-Wei Jang, Chia-Chun Hsu, Yi-Chen Yen, Yi-Ling Liu, Tsung-Hsien Chuang, Ssu-Han Wang, Yu-Ke Fu, Ching-Chuan Kuo, and et al. 2020. "ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line" Cancers 12, no. 1: 61. https://doi.org/10.3390/cancers12010061
APA StyleChen, Y. -L., Liu, K. -J., Jang, C. -W., Hsu, C. -C., Yen, Y. -C., Liu, Y. -L., Chuang, T. -H., Wang, S. -H., Fu, Y. -K., Kuo, C. -C., & Chen, Y. -W. (2020). ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers, 12(1), 61. https://doi.org/10.3390/cancers12010061