XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-Cell Lymphoma

 , , , , , , ,
, , , , , , , 
Abstract
:Simple Summary
Abstract

1. Introduction
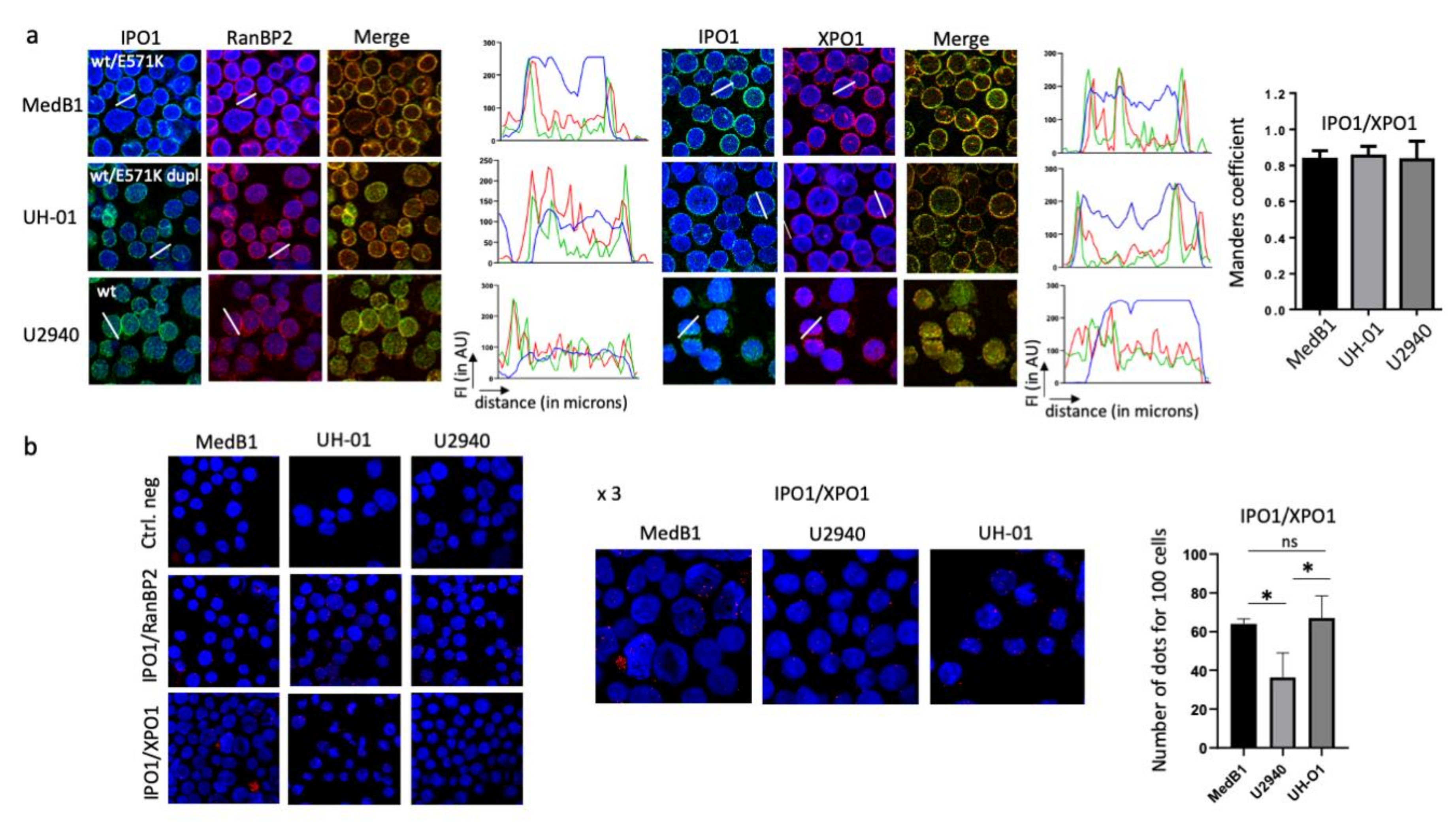
2. Results
2.1. PMBL and cHL Parental and Edited Cell Lines Display Various XPO1 Status
2.2. XPO1E571K Mutation is Present at the mRNA and Protein Levels
2.3. Nuclear Export Function of XPO1 is Maintained in MedB1 Cells
2.4. Wild-Type and Mutant XPO1 Proteins Localised in Different Compartments in PMBL Cells
2.5. Mutant and Wild-Type XPO1 Possess Similar Interactomes
2.6. XPO1E571K Protein Binds the Karyopherin β1 at the Outer Nuclear Membrane
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Cell Viability Assay
4.3. Indirect Immunofluorescence and Confocal Microscopy
4.4. Proximity Ligation Assay
4.5. Western Blotting
4.6. Immunoprecipitation, Mass Spectrometry and Protein Characterisation
4.7. CRISPR–Cas9 Editing
4.8. DNA and RNA Extraction, RT-PCR and Sanger Sequencing
4.9. DNA and RNA Pyrosequencing
4.10. Next-Generation Sequencing
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–962. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Monti, S.; Kutok, J.L.; Cattoretti, G.; Neuberg, D.; De Leval, L.; Kurtin, P.; Dal Cin, P.; Ladd, C.; Feuerhake, F.; et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B493 cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steidl, C.; Gascoyne, R.D. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood 2011, 118, 2659–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardin, F.; Pujals, A.; Pelletier, L.; Bohers, E.; Camus, V.; Mareschal, S.; Dubois, S.; Sola, B.; Ochmann, M.; Lemonnier, F.; et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am. J. Hematol. 2016, 91, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Viailly, P.J.; Mareschal, S.; Bohers, E.; Bertrand, P.; Ruminy, P.; Maingonnat, C.; Jais, J.P.; Peyrouze, P.; Figeac, M.; et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: A LYSA study. Clin. Cancer Res. 2016, 22, 2919–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, V.; Miloudi, H.; Taly, A.; Sola, B.; Jardin, F. XPO1 in B cell hematological malignancies: From recurrent somatic mutations to targeted therapy. J. Hematol. Oncol. 2017, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2012, 475, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Cosson, A.; Chapiro, E.; Bougacha, N.; Lambert, J.; Herbi, L.; Cung, H.A.; Algrin, C.; Keren, B.; Damm, F.; Gabillaud, C.; et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: Analysis of the central role of XPO1. Leukemia 2017, 31, 1625–1629. [Google Scholar] [CrossRef]
- Dai, H.; Ehrentraut, S.; Nagel, S.; Eberth, S.; Pommerenke, C.; Dirks, W.G.; Geffers, R.; Kalavalapalli, S.; Kaufmann, M.; Meyer, C.; et al. Genomic landscape of primary mediastinal B-cell lymphoma cell lines. PLoS ONE 2015, 10, e0139663. [Google Scholar] [CrossRef]
- Quentmeier, H.; Pommerenke, C.; Dirks, W.G.; Eberth, S.; Koeppel, M.; MacLeod, E.A.F.; Nagel, S.; Steube, K.; Uphoff, C.C.; Drexler, H.G. The LL-100 panel: 100 cell lines for blood cancer studies. Sci. Rep. 2019, 9, 8218. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Abdul Razak, F.R.; Terpstra, M.; Chan, F.C.; Saber, A.; Nijland, M.; van Imhoff, G.; Visser, L.; Gascoyne, R.; Steidl, C.; et al. The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia 2014, 28, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Neggers, J.E.; Vercruysse, T.; Jacquemyn, M.; Vanstreels, E.; Baloglu, E.; Shacham, S.; Crochiere, M.; Landesman, Y.; Daelemans, D. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem. Biol. 2015, 22, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xu-Monette, Z.Y.; Li, L.; Manyam, G.C.; Visco, C.; Tzankov, A.; Wang, J.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; et al. RelA NF-κB subunit activation as a therapeutic target in diffuse large B-cell lymphoma. Aging 2016, 8, 3321–3340. [Google Scholar] [CrossRef] [Green Version]
- Ritterhoff, T.; Das, H.; Hofhaus, G.; Schröder, R.R.; Flotho, A.; Melchior, F. The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a disassembly machine for Crm1-dependent nuclear export complexes. Nat. Commun. 2016, 7, 11482. [Google Scholar] [CrossRef] [Green Version]
- Kuusisto, H.V.; Wagstaff, K.M.; Alvisi, G.; Roth, D.M.; Jans, D.A. Global enhancement of nuclear localization-dependent nuclear transport in transformed cells. FASEB J. 2012, 26, 1181–1193. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Kırlı, K.; Karaca, S.; Dehne, H.J.; Samwer, M.; Ting Pan, K.; Lenz, C.; Urlaub, H.; Görlich1, D. A deep proteomics perspective on CRM1- mediated nuclear export and nucleocytoplasmic partitioning. eLife 2015, 4, e11466. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Berzginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. Human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Santisteban, I.; Arregi, I.; Alonso-Mariño, M.; Urbaneja, M.A.; Garcia-Vallejo, J.J.; Bañuelos, S.; Rodríguez, J.A. A cellular reporter to evaluate CRM1 nuclear export activity: Functional analysis of the cancer-related mutant E571K. Cell. Mol. Life Sci. 2016, 73, 4685–4699. [Google Scholar] [CrossRef]
- Taylor, J.; Sendino, M.; Gorelick, A.N.; Pastore, A.; Chang, M.T.; Penson, A.V.; Gavrila, E.I.; Stewart, C.; Melnik, E.M.; Herrejon Chavez, F.; et al. Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov. 2019, 9, 1452–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, M.; Chang, C.W.; Róna, G.; Smith, K.M.; Stewart, A.G.; Takeda, A.A.; Fontes, M.R.; Stewart, M.; Vértessy, B.G.; Forwood, J.K.; et al. Structural biology and regulation of protein import into the nucleus. J. Mol. Biol. 2016, 428, 2060–2090. [Google Scholar] [CrossRef]
- Hamada, M.; Haeger, A.; Jeganathan, K.B.; van Ree, J.H.; Malureanu, L.; Wälde, S.; Joseph, J.; Kehlenbach, R.H.; van Deursen, J.M. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J. Cell Biol. 2011, 194, 597–612. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Stark, L.A.; Dunlop, M.G. Nucleolar sequestration of RelA (p65) regulates NF-κB-driven transcription and apoptosis. Mol. Cell Biol. 2005, 25, 5985–6004. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Zhan, H.; Wang, G.; Li, S.; Cong, S.; Luo, Y.; Zhang, B. KPNB1, XPO7 and IPO8 mediate the translocatopn of NF-κB/p65 into the nucleus. Traffic 2013, 14, 1132–1143. [Google Scholar]
- Staudt, L.M. Oncogenic activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Stewart, M. Structural basis for the assembly of a nuclear export complex. Nature 2004, 432, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Matsuura, Y.; Liu, S.M.; Stewart, M. Structural basis for nuclear import complex dissociation by RanGTP. Nature 2005, 435, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.Y.; Chook, Y.M. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin. Cancer Biol. 2014, 27, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Biswas, A.; Süel, K.E.; Jackson, L.K.; Martinez, R.; Gu, H.; Chook, Y.M. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 2009, 458, 1136–1141. [Google Scholar] [CrossRef]
- Monecke, T.; Güttler, T.; Neumann, P.; Dickmanns, A.; Görlich, D.; Ficner, R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 2009, 324, 1087–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Port, S.A.; Monecke, T.; Dickmanns, A.; Spillner, C.; Hofele, R.; Urlaub, H.; Ficner, R.; Kehlenbach, R.H. Structural and functional characterization of CRM1-Nup214 interactions reveals multiple FG-binding sites involved in nuclear export. Cell Rep. 2015, 13, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Baumhardt, J.M.; Walker, J.S.; Lee, Y.; Shakya, B.; Brautigam, C.A.; Lapalombella, R.; Grishin, N.; Chook, Y.M. Recognition of nuclear export signals by CRM1 carrying the oncogenic E571K mutation. Mol. Biol. Cell 2020. [Google Scholar] [CrossRef]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 16, 61. [Google Scholar] [CrossRef]
- Nachmias, B.; Schimmer, A.D. Targeting nuclear import and export in hematological malignancies. Leukemia 2020. [Google Scholar] [CrossRef]
- He, S.; Miao, X.; Wu, Y.; Zhu, X.; Miao, X.; Yin, H.; He, Y.; Li, C.; Liu, Y.; Lu, X.; et al. Upregulation of nuclear transporter, Kpnβ1, contributes to accelerated cell proliferation- and cell adhesion-mediated drug resistance (CAM-DR) in diffuse large B-cell lymphoma. J. Cancer Res. Clin. Oncol. 2016, 142, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Li, R.; He, J.; Du, J.; Hou, J. Importin β1 mediates nuclear factor-κB signal transduction into the nuclei of myeloma cells and affects their proliferation and apoptosis. Cell Signal. 2015, 27, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Ritz, O.; Guiter, C.; Castellano, F.; Dorsch, K.; Melzner, J.; Jais, J.P. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood 2009, 114, 1236–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miloudi, H.; Leroy, K.; Jardin, F.; Sola, B. STAT6 is a cargo of exportin 1: Biological relevance in primary mediastinal B-cell lymphoma. Cell Signal. 2018, 46, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Body, S.; Esteve-Arenys, A.; Miloudi, H.; Recasens-Zorzo, C.; Tchakarska, G.; Moros, A.; Bustany, S.; Vidal-Crespo, A.; Rodriguez, V.; Lavigne, R.; et al. Cytoplasmic cyclin D1 controls the migration and invasiveness of mantle lymphoma cells. Sci. Rep. 2017, 7, 13946. [Google Scholar] [CrossRef] [PubMed]
- Demarta Gatsi, C.; Rivkin, A.; Di Bartolo, V.; Peronet, R.; Ding, S.; Commere, P.H.; Guillonneau, F.; Bellalou, J.; Brûlé, S.; Abou Karam, P.; et al. Histamine releasing factor and elongation factor 1 alpha secreted via malaria parasites extracellular vesicles promote immune evasion by inhibiting specific T cell responses. Cell Microbiol. 2019, 21, e13021. [Google Scholar] [CrossRef]
- Guernet, A.; Mungamuri, S.K.; Cartier, D.; Sachidanandam, R.; Jayaprakash, A.; Adriouch, S.; Vezain, M.; Charbonnier, F.; Rohkin, G.; Coutant, S.; et al. CRISPR-Barcoding for intratumor genetic heterogeneity modeling and functional analysis of oncogenic driver mutations. Mol Cell. 2016, 63, 526–538. [Google Scholar] [CrossRef] [Green Version]
- Maitre, E.; Bertrand, P.; Maingonnat, C.; Viailly, P.J.; Wiber, M.; Naguib, D.; Salaün, V.; Cornet, E.; Damaj, G.; Sola, B.; et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget 2018, 9, 28866–28876. [Google Scholar] [CrossRef] [Green Version]
- Yuen, H.F.; Chan, K.K.; Grills, C.; Murray, J.T.; Platt-Higgins, A.; Eldin, O.S.; O’Byrne, K.; Janne, P.; Fennell, D.A.; Johnston, P.G.; et al. Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways. Clin Cancer Res. 2012, 18, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Yuen, H.F.; Chan, K.K.; Platt-Higgins, A.; Dakir, E.H.; Matchett, K.B.; Haggag, Y.A.; Jithesh, P.V.; Habib, T.; Faheem, A.; Dean, F.A.; et al. Ran GTPase promotes cancer progression via Met receptor-mediated downstream signaling. Oncotarget 2016, 7, 75854–75864. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| K1106 | |||
|---|---|---|---|
| GO Term | Biological Process | Count in Gene Set | False Discovery Rate |
| GO:0046907 | Intracellular protein transport | 7/836 | 8.40 × 10−4 |
| GO Term | Molecular Function | Count in Gene Set | False Discovery Rate |
| GO:0003924 | GTPase activity | 8/283 | 7.42 × 10−7 |
| GO Term | Cellular Component | Count in Gene Set | False Discovery Rate |
| GO:0031090 | Organelle membrane | 20/2828 | 1.66 × 10−7 |
| INTERPRO Prot. domains | Count in Gene Set | False Discovery Rate | |
| IPR001806 | Small GTPase superfamily | 7/171 | 6.80 × 10−7 |
| SMART Prot. domains | Count in Gene Set | False Discovery Rate | |
| SM00175 | Rab subfamily of GTPases | 5/62 | 1.58 × 10−6 |
| MedB1 | |||
| GO Term | Biological Process | Count in Gene Set | False Discovery Rate |
| GO:0046907 | Intracellular protein transport | 10/1390 | 2.20 × 10−3 |
| GO Term | Molecular Function | Count in Gene Set | False Discovery Rate |
| GO:0003924 | GTPase activity | 4/283 | 3.60 × 10−7 |
| GO Term | Cellular Component | Count in Gene Set | False Discovery Rate |
| GO:0031090 | Organelle membrane | 16/2828 | 2.84 × 10−6 |
| INTERPRO Prot. domains | Count in Gene Set | False Discovery Rate | |
| IPR001806 | Small GTPase superfamily | 3/163 | 4.10 × 10−3 |
| SMART Prot. domains | Count in Gene Set | False Discovery Rate | |
| SM00175 | Rab subfamily of GTPases | 3/62 | 1.10 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miloudi, H.; Bohers, É.; Guillonneau, F.; Taly, A.; Gibouin, V.C.; Viailly, P.-J.; Jego, G.; Grumolato, L.; Jardin, F.; Sola, B. XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-Cell Lymphoma. Cancers 2020, 12, 2829. https://doi.org/10.3390/cancers12102829
Miloudi H, Bohers É, Guillonneau F, Taly A, Gibouin VC, Viailly P-J, Jego G, Grumolato L, Jardin F, Sola B. XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-Cell Lymphoma. Cancers. 2020; 12(10):2829. https://doi.org/10.3390/cancers12102829
Chicago/Turabian StyleMiloudi, Hadjer, Élodie Bohers, François Guillonneau, Antoine Taly, Vincent Cabaud Gibouin, Pierre-Julien Viailly, Gaëtan Jego, Luca Grumolato, Fabrice Jardin, and Brigitte Sola. 2020. "XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-Cell Lymphoma" Cancers 12, no. 10: 2829. https://doi.org/10.3390/cancers12102829
APA StyleMiloudi, H., Bohers, É., Guillonneau, F., Taly, A., Gibouin, V. C., Viailly, P. -J., Jego, G., Grumolato, L., Jardin, F., & Sola, B. (2020). XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-Cell Lymphoma. Cancers, 12(10), 2829. https://doi.org/10.3390/cancers12102829