Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
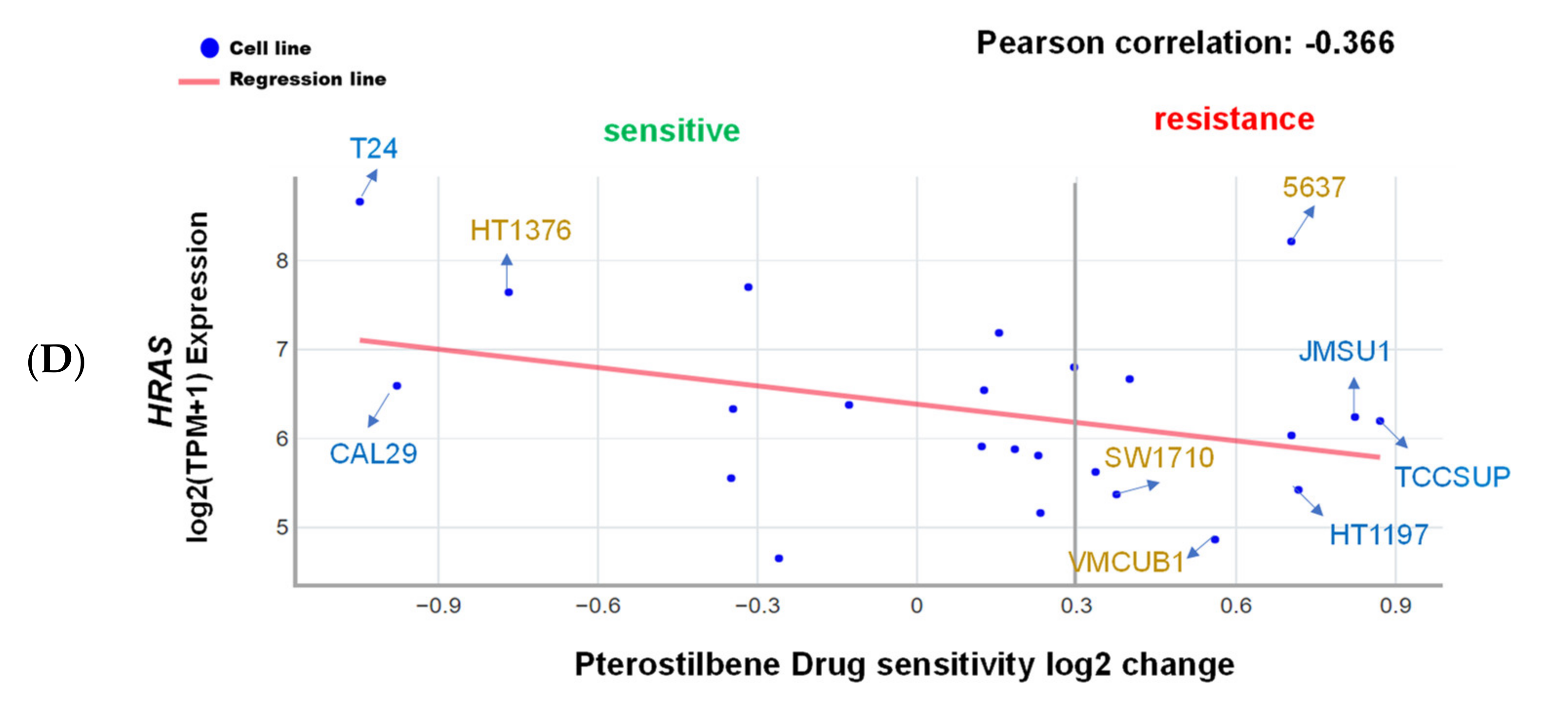
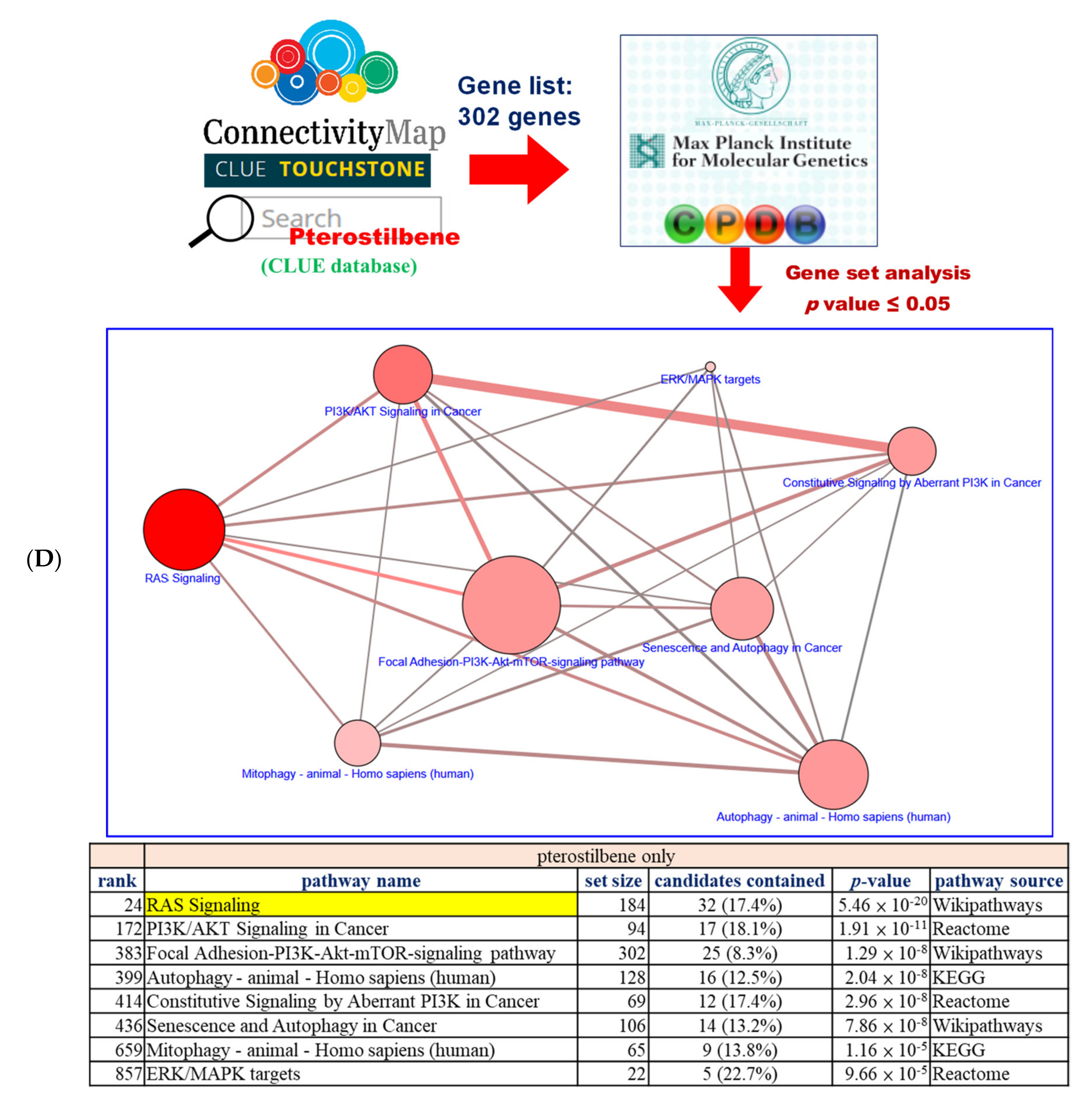
2.1. Pterostilbene Was Predicted to Sensitize Cisplatin-Resistant Bladder Cancer Cells with HRAS Gene Alterations via RAS-Related Autophagy and Cell Senescence Pathways
2.2. Pterostilbene-Enhanced Cytotoxic Response to Food and Drug Administration (FDA)-Approved Anticancer Drugs Was Indeed Associated with an Induction of Autophagy
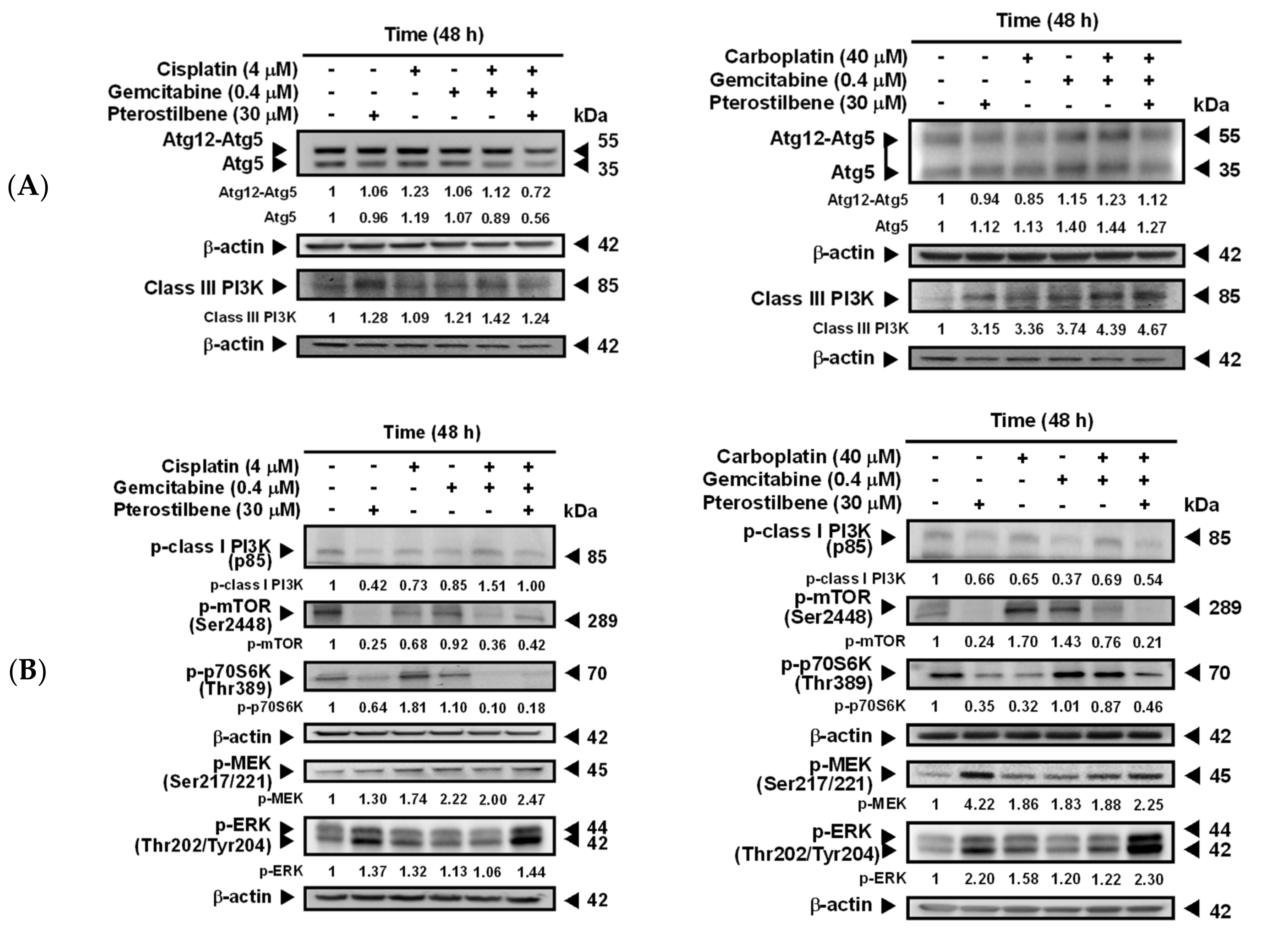
2.3. Pterostilbene-Induced Autophagy in T24 Cells Was Paralleled by an Activation of MEK/ERK and Class III PI3K, as Well as an Inhibition of Class I PI3K/mTOR/p70S6K
2.4. Pterostilbene-Induced Senescence and Cytotoxic Autophagy in T24 Cells
2.5. Pterostilbene Strengthened the Cisplatin/Gemcitabine-Induced Cytotoxicity and Reduced Tumorigenesis in T24 Xenograft-Bearing Mice without Exhibiting Physiologically Significant Side Effects
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Analysis of Mutation Frequency and Expression of the Genes
4.3. Regression Test
4.4. Cytotoxicity Assay
4.5. Flow Cytometric Analysis
4.6. Western Blot Analysis
4.7. Senescence Assay
4.8. Animal Study
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AVOs | acidic vesicular organelles |
| BUN | blood urea nitrogen |
| CCLE | Cancer Cell Line Encyclopedia |
| CLUE | C-Map and LINCS Unified Environment |
| C-Map | Connectivity Map |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| CPDB | ConsensusPathDB |
| CTRP | Cancer Therapeutics Response Portal |
| DepMap | Dependency Map |
| DMEM | Dulbecco’s modified Eagle medium |
| DMSO | dimethyl sulfoxide |
| DRIVE | Deep RNAi Interrogation of Viability Effects |
| ES | enrichment statistic |
| FDA | Food and Drug Administration |
| GDC | Genomic Data Commons |
| GOT | glutamate oxaloacetate transaminase |
| GPT | glutamate pyruvate transaminase |
| LINCS | Library of Integrated Network-Based Cellular Signatures |
| MFI | median fluorescence intensity |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide |
| PRISM | Profiling Relative Inhibition Simultaneously in Mixtures |
| SEMs | standard errors of the means |
| TCGA | The Cancer Genome Atlas |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeegers, M.P.; Goldbohm, R.A.; van den Brandt, P.A. A prospective study on active and environmental tobacco smoking and bladder cancer risk (The Netherlands). Cancer Causes Control 2002, 13, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Sastry, J.; Kellie, S.J. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr. Hematol. Oncol. 2005, 22, 441–445. [Google Scholar] [CrossRef]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Bajorin, D.F.; Rosenberg, J.E. First-line treatment and prognostic factors of metastatic bladder cancer for platinum-eligible patients. Hematol. Oncol. Clin. N. Am. 2015, 29, 319–328; ix–x. [Google Scholar] [CrossRef]
- Knickelbein, K.; Zhang, L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y. Bladder cancer and genetic mutations. Cell Biochem. Biophys. 2015, 73, 65–69. [Google Scholar] [CrossRef]
- Sugita, S.; Enokida, H.; Yoshino, H.; Miyamoto, K.; Yonemori, M.; Sakaguchi, T.; Osako, Y.; Nakagawa, M. HRAS as a potential therapeutic target of salirasib RAS inhibitor in bladder cancer. Int. J. Oncol. 2018, 53, 725–736. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Lin, H.S.; Yue, B.D.; Ho, P.C. Determination of pterostilbene in rat plasma by a simple HPLC-UV method and its application in pre-clinical pharmacokinetic study. Biomed. Chromatogr. 2009, 23, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Pai, R.S. Recent advances of resveratrol in nanostructured based delivery systems and in the management of HIV/AIDS. J. Control. Release 2014, 194C, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Crowell, J.A.; Korytko, P.J.; Morrissey, R.L.; Booth, T.D.; Levine, B.S. Resveratrol-associated renal toxicity. Toxicol. Sci. 2004, 82, 614–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. (Phila) 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [Green Version]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Hougee, S.; Faber, J.; Sanders, A.; de Jong, R.B.; van den Berg, W.B.; Garssen, J.; Hoijer, M.A.; Smit, H.F. Selective COX-2 inhibition by a Pterocarpus marsupium extract characterized by pterostilbene, and its activity in healthy human volunteers. Planta Med. 2005, 71, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Kapetanovic, I.M.; Muzzio, M.; Huang, Z.; Thompson, T.N.; McCormick, D.L. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother. Pharmacol. 2011, 68, 593–601. [Google Scholar] [CrossRef] [Green Version]
- McCormack, D.; McFadden, D. Pterostilbene and cancer: Current review. J. Surg. Res. 2012, 173, e53–e61. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. The ras gene family and human carcinogenesis. Mutat. Res. Rev. Genet. Toxicol. 1988, 195, 255–271. [Google Scholar] [CrossRef]
- Magne, N.; Fischel, J.L.; Tiffon, C.; Formento, P.; Dubreuil, A.; Renee, N.; Formento, J.L.; Francoual, M.; Ciccolini, J.; Etienne, M.C.; et al. Molecular mechanisms underlying the interaction between ZD1839 (‘Iressa’) and cisplatin/5-fluorouracil. Br. J. Cancer 2003, 89, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; Chuang, J.J.; Chang, T.Y.; Won, S.J.; Tsai, H.W.; Lee, C.T.; Cheng, H.L.; Tzai, T.S.; Liu, H.S.; Chow, N.H. Antiangiogenesis as the novel mechanism for justicidin A in the anticancer effect on human bladder cancer. Anticancer Drugs 2015, 26, 428–436. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cuervo, A.M.; Seglen, P.O. Methods for monitoring autophagy from yeast to human. Autophagy 2007, 3, 181–206. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G. Autophagy and NF-κB: Fight for fate. Cytokine Growth Factor Rev. 2007, 18, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.Y.; Yu, Q.J.; Zhang, R.D.; Liu, B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int. J. Biochem. Cell Biol. 2011, 43, 1263–1266. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Chang, Y.P.; Tsai, C.C.; Huang, W.C.; Wang, C.Y.; Chen, C.L.; Lin, Y.S.; Kai, J.I.; Hsieh, C.Y.; Cheng, Y.L.; Choi, P.C.; et al. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. J. Biol. Chem. 2010, 285, 28715–28722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.K.; Canto, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.C.; Chen, C.T.; Baron, D.; Onder, T.T.; Loewer, S.; Almeida, S.; Weismann, C.M.; Xu, P.; Houghton, J.M.; Gao, F.B.; et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat. Cell Biol. 2011, 13, 1214–1223. [Google Scholar] [CrossRef] [Green Version]
- Baumann, K. Organelle dynamics: Inheritance for pluripotency. Nat. Rev. Mol. Cell Biol. 2011, 12, 690–691. [Google Scholar] [CrossRef]
- Hinata, N.; Shirakawa, T.; Zhang, Z.; Matsumoto, A.; Fujisawa, M.; Okada, H.; Kamidono, S.; Gotoh, A. Radiation induces p53-dependent cell apoptosis in bladder cancer cells with wild-type-p53 but not in p53-mutated bladder cancer cells. Urol. Res. 2003, 31, 387–396. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Qi, Z.; Yin, S.; Zhang, N.; Liu, Y.; Liu, M.; Meng, J.; Zang, R.; Zhang, Z.; et al. The negative interplay between Aurora A/B and BRCA1/2 controls cancer cell growth and tumorigenesis via distinct regulation of cell cycle progression, cytokinesis, and tetraploidy. Mol. Cancer 2014, 13, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andresson, T.; Ruderman, J.V. The kinase Eg2 is a component of the Xenopus oocyte progesterone-activated signaling pathway. EMBO J. 1998, 17, 5627–5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, S.; Sakashita, G.; Ban, R.; Nagasawa, M.; Matsuzaki, H.; Murata, Y.; Taniguchi, H.; Shima, H.; Furukawa, K.; Urano, T. Phospho-regulation of human protein kinase Aurora-A: Analysis using anti-phospho-Thr288 monoclonal antibodies. Oncogene 2006, 25, 7691–7702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.T.; Cao, L.; Chen, B.P.; Cheng, W.H. Selenoprotein H suppresses cellular senescence through genome maintenance and redox regulation. J. Biol. Chem. 2014, 289, 34378–34388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, N.K.; Krishnasamy, M.; Isenring, E.A. The effect of nutrition intervention in lung cancer patients undergoing chemotherapy and/or radiotherapy: A systematic review. Nutr. Cancer 2014, 66, 47–56. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.X.; Ouyang, L.; Cheng, Y.; Liu, B. Plant natural compounds: Targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif. 2012, 45, 466–476. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Lin, C.W.; Yang, S.F.; Sheu, G.T.; Yu, Y.Y.; Chen, M.K.; Chiou, H.L. A combination of pterostilbene with autophagy inhibitors exerts efficient apoptotic characteristics in both chemosensitive and chemoresistant lung cancer cells. Toxicol. Sci. 2014, 137, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.J.; Tsai, S.J.; Ho, C.T.; Pan, M.H.; Ho, Y.S.; Wu, C.H.; Wang, Y.J. Chemopreventive effects of pterostilbene on urethane-induced lung carcinogenesis in mice via the inhibition of EGFR-mediated pathways and the induction of apoptosis and autophagy. J. Agric. Food Chem. 2012, 60, 11533–11541. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, L.; Wang, X.; Zhang, J.; Han, W.; Feng, L.; Sun, J.; Jin, H.; Wang, X.J. Pterostilbene simultaneously induces apoptosis, cell cycle arrest and cyto-protective autophagy in breast cancer cells. Am. J. Transl. Res. 2012, 4, 44–51. [Google Scholar]
- Chakraborty, A.; Bodipati, N.; Demonacos, M.K.; Peddinti, R.; Ghosh, K.; Roy, P. Long term induction by pterostilbene results in autophagy and cellular differentiation in MCF-7 cells via ROS dependent pathway. Mol. Cell. Endocrinol. 2012, 355, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Ho, C.T.; Wang, Y.J. Pterostilbene induces autophagy and apoptosis in sensitive and chemoresistant human bladder cancer cells. Mol. Nutr. Food Res. 2010, 54, 1819–1832. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Reznikoff, C.A.; Belair, C.; Savelieva, E.; Zhai, Y.; Pfeifer, K.; Yeager, T.; Thompson, K.J.; DeVries, S.; Bindley, C.; Newton, M.A.; et al. Long-term genome stability and minimal genotypic and phenotypic alterations in HPV16 E7-, but not E6-, immortalized human uroepithelial cells. Genes Dev. 1994, 8, 2227–2240. [Google Scholar] [CrossRef] [Green Version]
- Mikula-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Ksiazek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Chen, Y.Y.; Yeh, Y.L.; Wang, Y.J.; Chen, R.J. Stilbene compounds inhibit tumor growth by the induction of cellular senescence and the inhibition of telomerase activity. Int. J. Mol. Sci. 2019, 20, 2716. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Slobodnyuk, K.; Radic, N.; Ivanova, S.; Llado, A.; Trempolec, N.; Zorzano, A.; Nebreda, A.R. Autophagy-induced senescence is regulated by p38alpha signaling. Cell Death Dis. 2019, 10, 376. [Google Scholar] [CrossRef]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, Y.; Jeong, J.H.; Ryu, J.H.; Kim, W.Y. ATM/CHK/p53 pathway dependent chemopreventive and therapeutic activity on lung cancer by pterostilbene. PLoS ONE 2016, 11, e0162335. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Wu, P.H.; Ho, C.T.; Way, T.D.; Pan, M.H.; Chen, H.M.; Ho, Y.S.; Wang, Y.J. P53-dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment. Cell Death Dis. 2017, 8, e2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.J.; Haluschak, J.J.; Johnson, D.; Schwartz, S.; Morrison, L.J.; Lippa, M.; Hatzivassiliou, G.; Tan, J. p53 mutations in bladder carcinoma cell lines. Oncol. Res. 1994, 6, 569–579. [Google Scholar]
- Makarov, E.M.; Shtam, T.A.; Kovalev, R.A.; Pantina, R.A.; Varfolomeeva, E.Y.; Filatov, M.V. The rare nonsense mutation in p53 triggers alternative splicing to produce a protein capable of inducing apoptosis. PLoS ONE 2017, 12, e0185126. [Google Scholar] [CrossRef] [Green Version]
- Giet, R.; Petretti, C.; Prigent, C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005, 15, 241–250. [Google Scholar] [CrossRef]
- Liu, H.S.; Ke, C.S.; Cheng, H.C.; Huang, C.Y.; Su, C.L. Curcumin-induced mitotic spindle defect and cell cycle arrest in human bladder cancer cells is partly through inhibition of Aurora-A. Mol. Pharmacol. 2011, 80, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Ke, C.S.; Liu, H.S.; Yen, C.H.; Huang, G.C.; Cheng, H.C.; Huang, C.Y.; Su, C.L. Curcumin-induced Aurora-A suppression not only causes mitotic defect and cell cycle arrest but also alters chemosensitivity to anticancer drugs. J. Nutr. Biochem. 2014, 25, 526–539. [Google Scholar] [CrossRef]
- Bastos, R.N.; Barr, F.A. Plk1 negatively regulates Cep55 recruitment to the midbody to ensure orderly abscission. J. Cell Biol. 2010, 191, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.T.; Ettinger, A.W.; Huttner, W.B.; Doxsey, S.J. Resurrecting remnants: The lives of post-mitotic midbodies. Trends Cell Biol. 2013, 23, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; Li, W.M.; Wu, W.J.; Chai, C.Y.; Chang, T.Y.; Sun, Y.; Cheng, C.J.; Shiue, Y.L.; Su, S.J.; Cheng, H.L.; et al. Epithelial membrane protein 2 is a prognostic indictor for patients with urothelial carcinoma of the upper urinary tract. Am. J. Pathol. 2013, 183, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, C.H.; Su, C.L.; Huang, C.W.; Liu, H.S.; Lin, C.N.; Won, S.J. Justicidin A decreases the level of cytosolic Ku70 leading to apoptosis in human colorectal cancer cells. Carcinogenesis 2005, 26, 1716–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.L.; Wang, Y.T.; Chang, M.H.; Fang, K.; Chen, K. The novel heterocyclic Trioxirane [(1,3,5-Tris Oxiran-2-yl)Methyl)-1,3,5-Triazinane-2,4,6-Trione (TATT)] exhibits a better anticancer effect than platinum-based chemotherapy by induction of apoptosis and curcumin further enhances its chemosensitivity. Cell Biochem. Biophys. 2014, 68, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yen, C.H.; Liu, H.S.; Wu, S.Y.; Lan, S.H.; Jiang-Shieh, Y.F.; Lin, C.N.; Su, C.L. Justicidin A-induced autophagy flux enhances apoptosis of human colorectal cancer cells via class III PI3K and Atg5 pathway. J. Cell. Physiol. 2015, 230, 930–946. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Al Jameil, N.; Arjumand, S.; Khan, M.F.; Tabassum, H.; Alenzi, N.; Hijazy, S.; Alenzi, S.; Subaie, S.; Fatima, S. Comparative study of serum copper, iron, magnesium, and zinc in type 2 diabetes-associated proteinuria. Biol. Trace Elem. Res. 2015, 168, 321–329. [Google Scholar] [CrossRef]
- Webster, R.M. Combination therapies in oncology. Nat. Rev. Drug Discov. 2016, 15, 81–82. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Rare genetic diseases with defects in DNA repair: Opportunities and challenges in orphan drug development for targeted cancer therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef] [Green Version]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-T.; Huang, Z.-Y.; Tang, H.-H.; Kuo, W.-T.; Wu, S.-Y.; Lan, S.-H.; Chang, K.-H.; Lin, P.-L.; Lee, M.-F.; Cheng, H.-C.; et al. Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS. Cancers 2020, 12, 2869. https://doi.org/10.3390/cancers12102869
Chen Y-T, Huang Z-Y, Tang H-H, Kuo W-T, Wu S-Y, Lan S-H, Chang K-H, Lin P-L, Lee M-F, Cheng H-C, et al. Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS. Cancers. 2020; 12(10):2869. https://doi.org/10.3390/cancers12102869
Chicago/Turabian StyleChen, Yi-Ting, Zi-Yi Huang, Han-Hsuan Tang, Wan-Ting Kuo, Shan-Ying Wu, Sheng-Hui Lan, Kai-Hsun Chang, Pin-Lun Lin, Ming-Fen Lee, Hung-Chi Cheng, and et al. 2020. "Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS" Cancers 12, no. 10: 2869. https://doi.org/10.3390/cancers12102869
APA StyleChen, Y. -T., Huang, Z. -Y., Tang, H. -H., Kuo, W. -T., Wu, S. -Y., Lan, S. -H., Chang, K. -H., Lin, P. -L., Lee, M. -F., Cheng, H. -C., Liu, H. -S., Huang, C. -Y. F., Huang, G. -C., & Su, C. -L. (2020). Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS. Cancers, 12(10), 2869. https://doi.org/10.3390/cancers12102869