Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
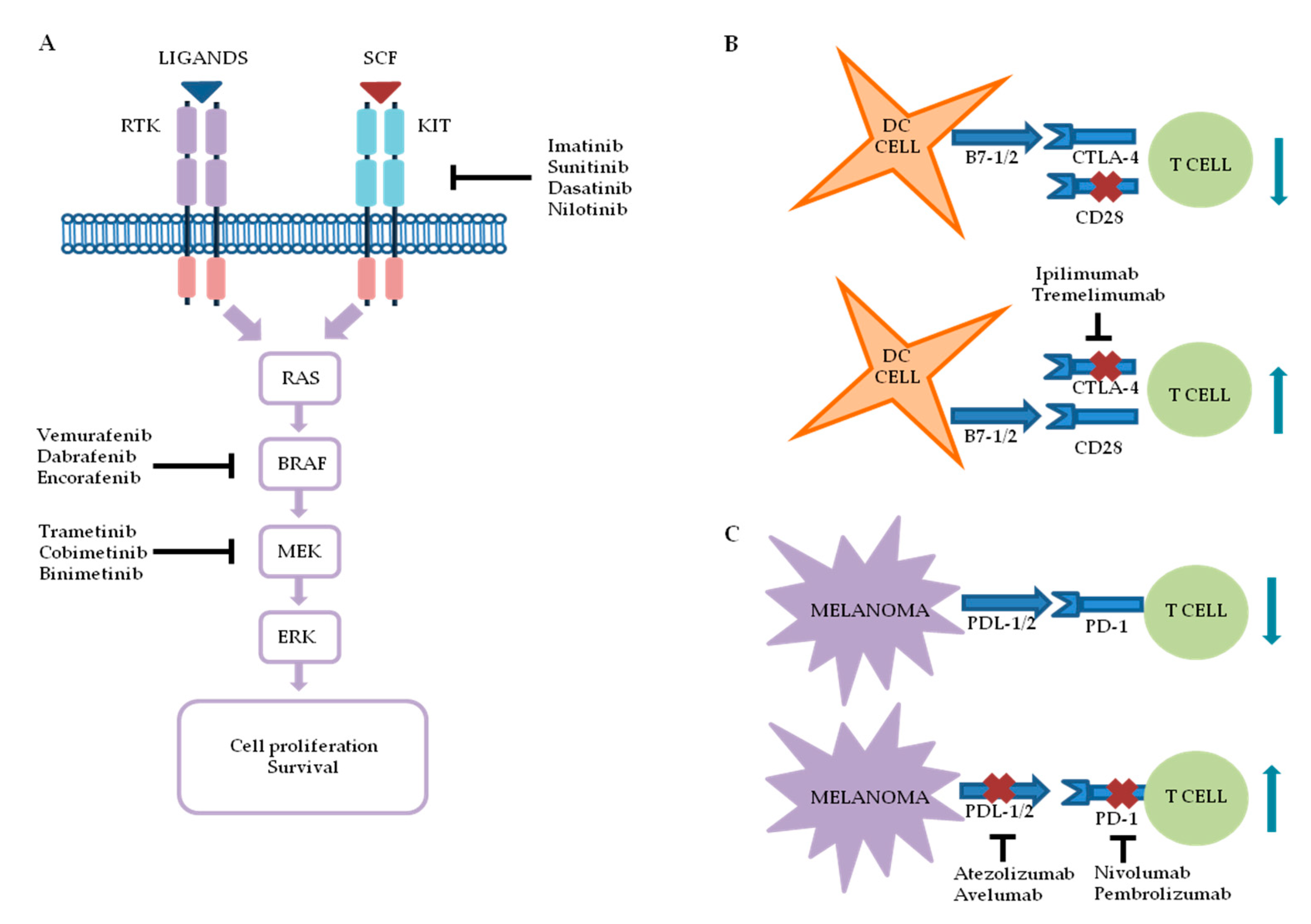
2. Melanoma Targeted Therapy and Immunotherapy: An Overview
2.1. Targeted Therapy
2.1.1. BRAF Inhibitors
2.1.2. Mitogen-Activated Protein Kinase Kinase (MEK) Inhibitors
2.1.3. V-kit Hardy–Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog (KIT) Inhibitors
2.2. Immunotherapy
2.2.1. Anti-CTLA-4
2.2.2. Anti-PD-1
2.2.3. Alternative Melanoma Immunotherapies
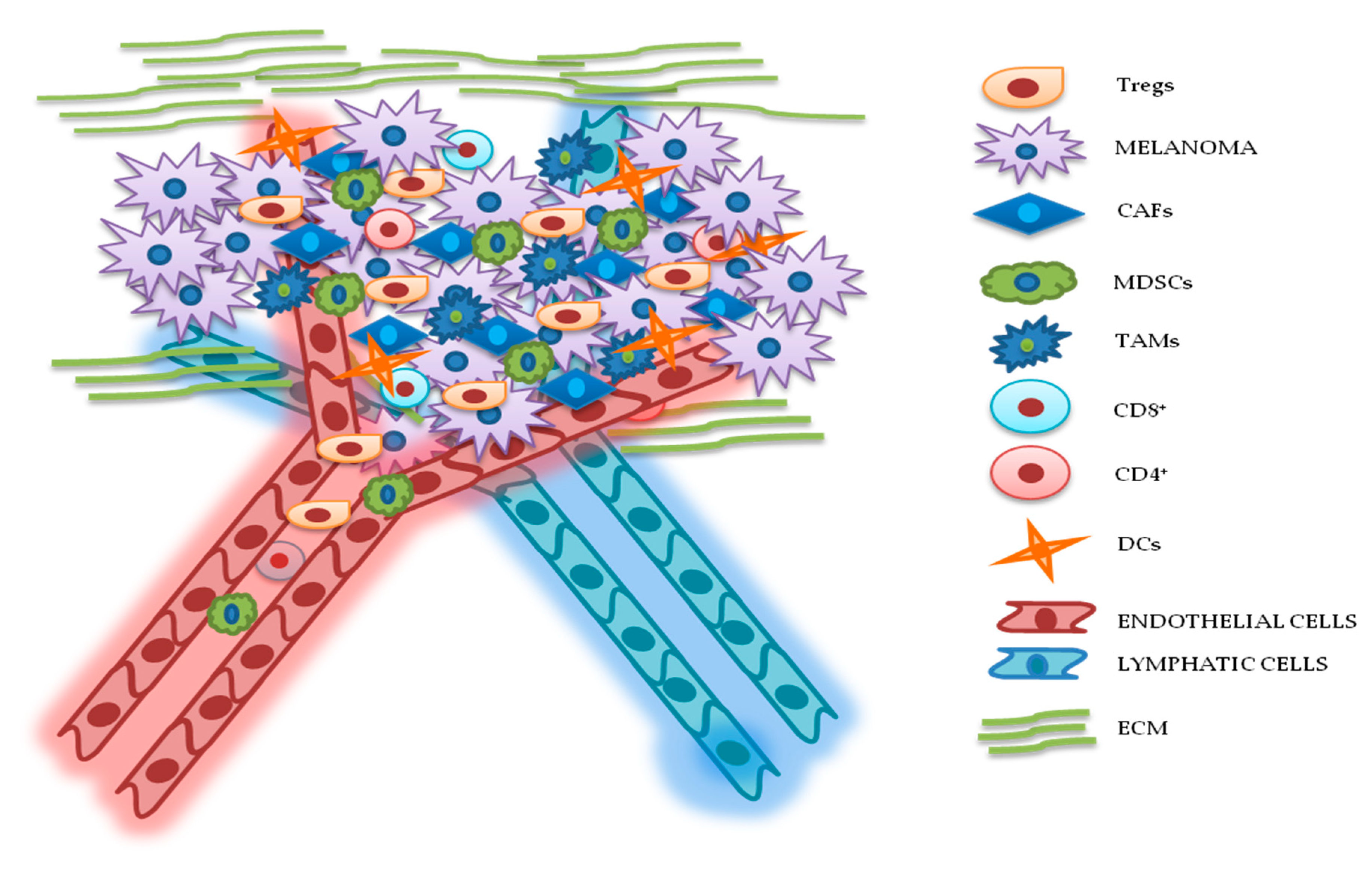
3. TME Implications in Drug Resistance for Melanoma
3.1. Cellular Components
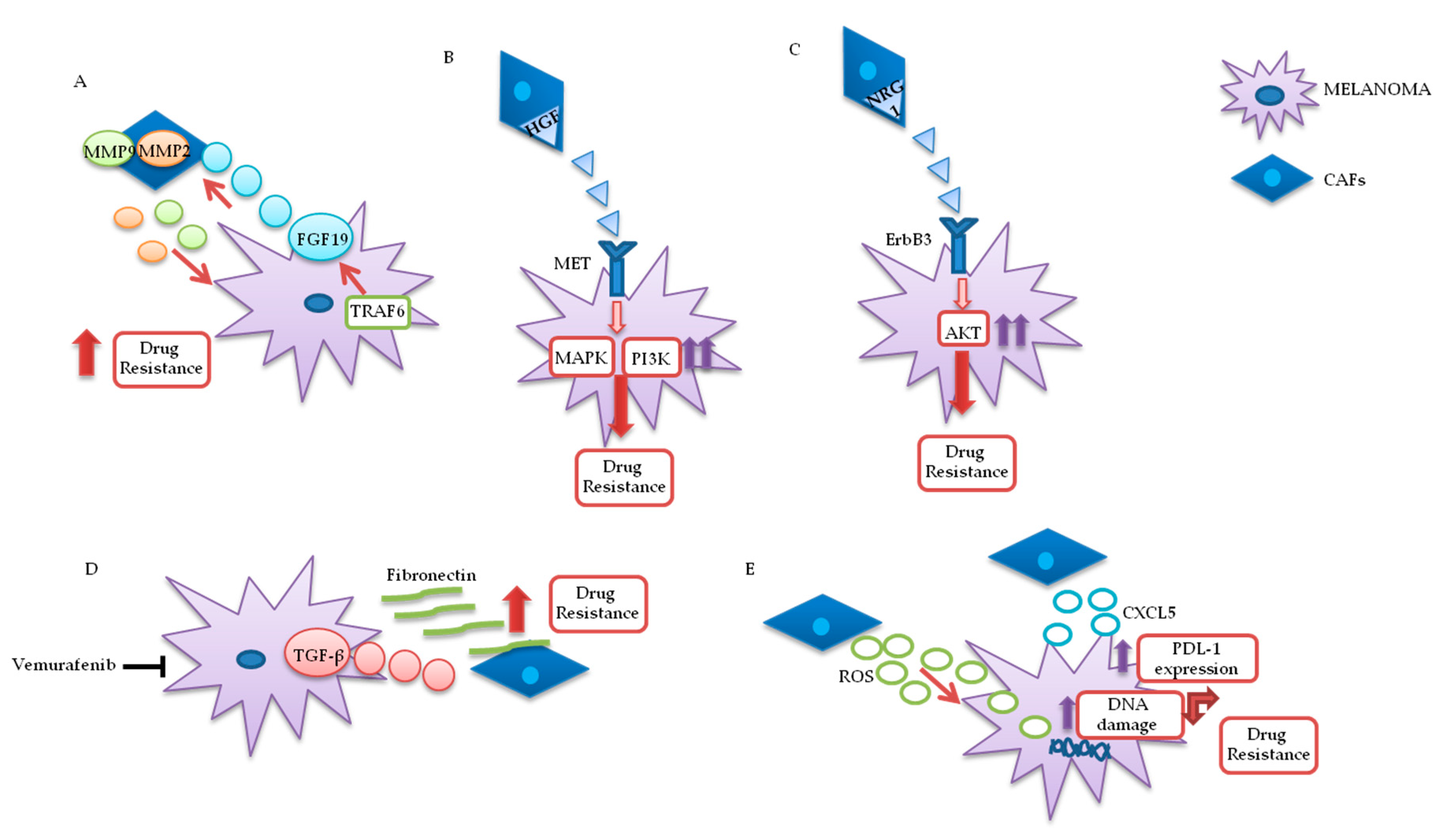
3.1.1. Cancer-Associated Fibroblasts (CAFs)
3.1.2. Lymphocytes
3.1.3. MDSCs
3.1.4. Tumor-Associated Macrophages (TAMs)
3.1.5. DCs
3.2. ECM
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| APC | Antigen presenting cell |
| APE1 | Apurinic/apyrimidinic endonuclease 1 |
| BAD | BCL2 associated agonist of cell death |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| CAFs | Cancer-associated fibroblasts |
| CAR-T | Chimeric antigen receptor T-cells |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CCL-22 | C-C motif chemokine 22 |
| CD | Cluster differentiation |
| CRAF | v-raf1 murine leukemia viral oncogene homolog 1 |
| CTL | Cytotoxic lymphocytes |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL-2 | CXC chemokine ligand 2 |
| CXCL5 | CXC motif chemokine 5 |
| DCs | Dendritic cells |
| DNAM-1 | DNAX accessory molecule-1 |
| ECM | Extracellular matrix |
| EMT | Epithelial mesenchymal transition |
| ErbB3 | V-erb-b2 avian erythroblastic leukemia viral oncogene homolog3 |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| FAP | Fibroblast activation protein |
| FcγR | Fragment crystallizable-gamma receptor |
| FDA | Food and Drug Administration |
| FGF 2-19 | Fibroblast growth factor 2-19 |
| FGFR-3 | Fibroblast growth factor receptor 3 |
| FSP1 | Fibroblast specific protein 1 |
| GSK3β | Glycogen synthase kinase 3 beta |
| Gp | Glycoprotein |
| HER3 | V-erb-b2 avian erythroblastic leukemia viral oncogene homolog 3 |
| HGF | Hepatocyte growth factor |
| ICIs | Immune checkpoint inhibitors |
| IDO | Indoleamine-2,3-dioxygenase |
| Ig | Immunoglobulin |
| IGF-1 | Insulin-like growth factor 1 |
| IGF-1R | Insulin-like growth factor receptor 1 |
| IL | Interleukin |
| ITIM | Immunoreceptor tyrosine-based inhibition motif |
| KIT | v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog |
| LIPA | Lysosomal acid lipase A |
| MAPK | Mitogen-activated protein kinase |
| MCL-1 | Myeloid leukemia cell protein 1 |
| MCH-I | Major histocompatibility complex 1 |
| MDSCs | Myeloid-derived suppressor cells |
| MEK | Mitogen-activated protein kinase kinase |
| MET | Mesenchymal epithelial transition receptor |
| MHC | Major histocompatibility complex |
| miRNA | microRNA |
| MITF | Microphthalmia-associated transcription factor |
| MMPs | Matrix metalloproteinases |
| NFkB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGS | Next-generation sequencing |
| NK | Natural killer |
| NKG2D | Natural killer group 2D |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| NRG-1 | Neuregulin 1 |
| ORR | Objective response rate |
| OS | Overall survival |
| PBMC | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death protein 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PDL-1/2 | Programmed death ligands 1/2 |
| PI3K | Phosphatidyl Inositol 3-kinase |
| PFS | Progression-free survival |
| PTEN | Phosphatase and tensin homolog on chromosome 10 |
| ROS | Reactive oxygen species |
| RTK | Tyrosine kinase receptor |
| sFRP2 | Secreted frizzled-related protein 2 |
| STAT | Signal transducer and activator of transcription |
| SRC | V-src sarcoma (Schmidt–Ruppin A-2) viral oncogene homolog avian |
| TAB | Tumor-associated B cells |
| TAMs | Tumor-associated macrophages |
| TCF7 | Transcription factor 7 |
| TGF-β | Transforming growth factor β |
| TIGIT | T cell Ig and ITIM domain |
| TIL | Tumor-infiltrating lymphocytes |
| Tim3 | Immunoglobulin and mucin domain-containing molecule 3 |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor α |
| TRAF6 | Tumor necrosis factor receptor-associated factor 6 |
| Tregs | Regulatory T cells |
| VEGF | Vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T cell activation |
| Wnt | wingless type MMTV integration site family member |
| α-SMA | α smooth muscle actin |
References
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.C. REVIEW ARTICLE: How to make a melanoma: What do we know of the primary clonal events? Pigment. Cell Melanoma Res. 2007, 21, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Sanlorenzo, M.; Vujic, I.; Posch, C.; Dajee, A.; Yen, A.; Kim, S.; Ashworth, M.; Rosenblum, M.D.; Algazi, A.; Osella-Abate, S.; et al. Melanoma immunotherapy. Cancer Biol. Ther. 2014, 15, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.; Arance, A.; Mandalà, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsová, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Saito, M.; Momma, T.; Kono, K. Targeted therapy according to next generation sequencing-based panel sequencing. FUKUSHIMA J. Med Sci. 2018, 64, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Inhibitors: Rationale and Importance to Inhibiting These Pathways in Human Health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma Cell Death Mechanisms. Chonnam Med J. 2018, 54, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, V.; Ramelyte, E.; Thurneysen, S.; Pitocco, R.; Bentele-Jaberg, N.; Goldinger, S.; Dummer, R.; Mangana, J. Developments in targeted therapy in melanoma. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 581–593. [Google Scholar] [CrossRef]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Gray, D.C.; Eby, M.T.; Tien, J.Y.; Wong, L.; Bower, J.; Gogineni, A.; Zha, J.; Cole, M.J.; Stern, H.M.; et al. Oncogenic BRAF Is Required for Tumor Growth and Maintenance in Melanoma Models. Cancer Res. 2006, 66, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Jacobetz, M.A.; Robertson, G.P.; Herlyn, M.; Tuveson, D.A. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003, 63, 5198–5202. [Google Scholar]
- Hoeflich, K.P.; Jaiswal, B.; Davis, D.P.; Seshagiri, S. Inducible BRAF Suppression Models for Melanoma Tumorigenesis. Methods Enzymol. 2008, 439, 25–38. [Google Scholar] [CrossRef]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.K.; Zarzoso, I.; Daud, A.I. PD-1 and PD-L1 antibodies for melanoma. Hum. Vaccines Immunother. 2014, 10, 3111–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, J.; Stenger, M. Dabrafenib in advanced melanoma with BRAF V600E mutation. J. Community Support. Oncol. 2014, 12, 48–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Morales, D.; Lombart, F.; Truchot, A.; Maire, P.; Hussein, M.; Hamitou, W.; Vigneron, P.; Galmiche, A.; Lok, C.; Vayssade, M. 3D Coculture Models Underline Metastatic Melanoma Cell Sensitivity to Vemurafenib. Tissue Eng. Part A 2019, 25, 1116–1126. [Google Scholar] [CrossRef]
- Delord, J.-P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.J.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF -Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [Green Version]
- Saei, A.; Eichhorn, P.J.A. Saei Adaptive Responses as Mechanisms of Resistance to BRAF Inhibitors in Melanoma. Cancers 2019, 11, 1176. [Google Scholar] [CrossRef] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.R.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.; Pritchard, C.A.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nat. Cell Biol. 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Del Curatolo, A.; Conciatori, F.; Incani, U.C.; Bazzichetto, C.; Falcone, I.; Corbo, V.; D’Agosto, S.L.; Eramo, A.; Sette, G.; Sperduti, I.; et al. Therapeutic potential of combined BRAF/MEK blockade in BRAF-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 2018, 37, 140. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nat. Cell Biol. 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojaniello, C.; Festino, L.; Vanella, V.; Ascierto, P.A. Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert Rev. Clin. Pharmacol. 2019, 12, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; Van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging Ras Back in the Ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Schadendorf, D.; A Ascierto, P.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandalà, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Munoz-Couselo, E.; Adelantado, E.Z.; Vélez, C.O.; García, J.S.; Perez-Garcia, J.M.; Ortiz, C. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [Green Version]
- Sarkisian, S.; Davar, D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des. Dev. Ther. 2018, 12, 2553–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F.; Azar, S.; et al. KIT Gene Mutations and Copy Number in Melanoma Subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handolias, D.; Salemi, R.; Murray, W.; Tan, A.; Liu, W.; Viros, A.; Dobrovic, A.; Kelly, J.; McArthur, G.A. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment. Cell Melanoma Res. 2010, 23, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Goldinger, S.M.; Murer, C.; Stieger, P.; Dummer, R. Targeted therapy in melanoma – the role of BRAF, RAS and KIT mutations. Eur. J. Cancer Suppl. 2013, 11, 92–96. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 28 September 2020).
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of melanoma. Współczesna Onkol. 2018, 22, 61–67. [Google Scholar] [CrossRef]
- Şimşek, M.; Tekin, S.B.; Bilici, M. Immunological Agents Used in Cancer Treatment. Eurasian J. Med. 2019, 51, 90–94. [Google Scholar] [CrossRef]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, E4223–E4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eroglu, Z.; Kim, D.W.; Wang, X.; Camacho, L.H.; Chmielowski, B.; Seja, E.; Villanueva, A.; Ruchalski, K.; Glaspy, J.A.; Kim, K.B.; et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur. J. Cancer 2015, 51, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A. Clinical Development of the Anti–CTLA-4 Antibody Tremelimumab. Semin. Oncol. 2010, 37, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, F.; Jiang, F.; Lv, X.; Zhang, R.; Lu, A.; Zhang, G. A Mini-Review for Cancer Immunotherapy: Molecular Understanding of PD-1/PD-L1 Pathway & Translational Blockade of Immune Checkpoints. Int. J. Mol. Sci. 2016, 17, 1151. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Johnston, R.J.; Lu, L.-S.; Han, M.; Thudium, K.; Yao, D.; Quigley, M.; Valle, J.; Wang, C.; et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLoS ONE 2016, 11, e0161779. [Google Scholar] [CrossRef] [Green Version]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Li, Y.; Tian, Y.; Zhang, H.; Cai, W.; Chen, A.; Chen, L.; Bao, Y.; Xiang, B.; Kan, H.; et al. High-affinity human programmed death-1 ligand-1 variant promotes redirected T cells to kill tumor cells. Cancer Lett. 2019, 447, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Simeone, E.; Ascierto, P.A. Anti-PD-1 and PD-L1 antibodies in metastatic melanoma. Melanoma Manag. 2017, 4, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Hamid, O.; Daud, A.I.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.F.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Hamid, O.; Molinero, L.; Bolen, C.R.; Sosman, J.A.; Muñoz-Couselo, E.; Kluger, H.M.; McDermott, D.F.; Powderly, J.D.; Sarkar, I.; Ballinger, M.; et al. Safety, Clinical Activity, and Biological Correlates of Response in Patients with Metastatic Melanoma: Results from a Phase I Trial of Atezolizumab. Clin. Cancer Res. 2019, 25, 6061–6072. [Google Scholar] [CrossRef] [Green Version]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.; Taylor, M.; Wyrwicz, L.; Lee, K.-W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Jean, M.; Knol, A.-C.; Volteau, C.; Quéreux, G.; Peuvrel, L.; Brocard, A.; Pandolfino, M.-C.; Saiagh, S.; Nguyen, J.-M.; Bedane, C.; et al. Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes in Advanced Melanoma Patients. J. Immunol. Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, B.; Uslu, U. CAR -T cell therapy in melanoma: A future success story? Exp. Dermatol. 2018, 27, 1315–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermont, A.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Vigneron, N.; Ooms, A.; Morel, S.; Ma, W.; DeGiovanni, G.; Eynde, B.J.V.D. A peptide derived from melanocytic protein gp100 and presented by HLA-B35 is recognized by autologous cytolytic T lymphocytes on melanoma cells. Tissue Antigens 2005, 65, 156–162. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Testori, A.; Richards, J.; Whitman, E.; Mann, G.B.; Lutzky, J.; Camacho, L.H.; Parmiani, G.; Tosti, G.; Kirkwood, J.M.; Hoos, A.; et al. Phase III Comparison of Vitespen, an Autologous Tumor-Derived Heat Shock Protein gp96 Peptide Complex Vaccine, With Physician’s Choice of Treatment for Stage IV Melanoma: The C-100-21 Study Group. J. Clin. Oncol. 2008, 26, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Tosti, G.; Di Pietro, A.; Ferrucci, P.F.; Testori, A. HSPPC-96 vaccine in metastatic melanoma patients: From the state of the art to a possible future. Expert Rev. Vaccines 2009, 8, 1513–1526. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, Y.-C.; Sun, C.; Chen, Q. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Wu, Z.; Jin, H.; Hashimoto, N.; Liu, T.; Phan, S.H. CCAAT/Enhancer-Binding Protein β Isoforms and the Regulation of α-Smooth Muscle Actin Gene Expression by IL-1β. J. Immunol. 2004, 173, 4661–4668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61. [Google Scholar] [CrossRef]
- Cornil, I.; Theodorescu, D.; Man, S.; Herlyn, M.; Jambrosic, J.; Kerbel, R.S. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc. Natl. Acad. Sci. USA 1991, 88, 6028–6032. [Google Scholar] [CrossRef] [Green Version]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J.; Smetana, K. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, K.; Wickett, R.R.; Kadekaro, A.L.; Zhang, Y. Targeted deactivation of cancer-associated fibroblasts by β-catenin ablation suppresses melanoma growth. Tumor Biol. 2016, 37, 14235–14248. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Zeng, W.; Zhang, J.; Cai, L.; Wu, Z.; Su, J.; Xiao, Y.; Liu, N.; Tang, L.; et al. TRAF6 Activates Fibroblasts to Cancer-Associated Fibroblasts through FGF19 in Tumor Microenvironment to Benefit the Malignant Phenotype of Melanoma Cells. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nat. Cell Biol. 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capparelli, C.; Rosenbaum, S.; Berger, A.C.; Aplin, A.E. Fibroblast-derived Neuregulin 1 Promotes Compensatory ErbB3 Receptor Signaling in Mutant BRAF Melanoma*. J. Biol. Chem. 2015, 290, 24267–24277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorenko, I.V.; Wargo, J.A.; Flaherty, K.T.; Messina, J.L.; Smalley, K.S. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J. Investig. Dermatol. 2015, 135, 3115–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nat. Cell Biol. 2016, 532, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Sakakura, K.; Kawabata-Iwakawa, R.; Rokudai, S.; Toyoda, M.; Nishiyama, M.; Chikamatsu, K. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1407–1417. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Ziani, L.; Ben Safta-Saadoun, T.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar]
- Flach, E.H.; Rebecca, V.W.; Herlyn, M.; Smalley, K.S.; Anderson, A.R. Fibroblasts Contribute to Melanoma Tumor Growth and Drug Resistance. Mol. Pharm. 2011, 8, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiago, M.; De Oliveira, E.M.; Brohem, C.A.; Pennacchi, P.C.; Paes, R.D.; Haga, R.B.; Campa, A.; Barros, S.B.D.M.; Smalley, K.S.; Maria-Engler, S.S. Fibroblasts Protect Melanoma Cells from the Cytotoxic Effects of Doxorubicin. Tissue Eng. Part A 2014, 20, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, A.; Bosselut, R. CD4⧸CD8 Coreceptors in Thymocyte Development, Selection, and Lineage Commitment: Analysis of the CD4⧸CD8 Lineage Decision. Adv. Immunol. 2004, 83, 91–131. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Cetintas, V.B.; Batada, N.N. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J. Transl. Med. 2020, 18, 45. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Richards, J.-A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2013, 33, 4632–4642. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2015, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Van Der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; Van Akkooi, A.C.; Braber, M.V.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy, Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef] [Green Version]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nat. Cell Biol. 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.H.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiol. 2017, 222, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Cristiani, C.M.; Garofalo, C.; Passacatini, L.C.; Carbone, E. New avenues for melanoma immunotherapy: Natural Killer cells? Scand. J. Immunol. 2020, 91, e12861. [Google Scholar] [CrossRef] [PubMed]
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. OncoImmunology 2017, 7, e1392426. [Google Scholar] [CrossRef]
- Kondělková, K.; Vokurková, D.; Krejsek, J.; Borska, L.; Fiala, Z.; Andrys, C. Regulatory T cells (Treg) and Their Roles in Immune System with Respect to Immunopathological Disorders. Acta Medica (Hradec Kralove, Czech Republic) 2010, 53, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Napolitano, M.; Celentano, E.; Simeone, E.; Gentilcore, G.; Daponte, A.; Capone, M.; Caracò, C.; Calemma, R.; Beneduce, G.; et al. Regulatory T cell frequency in patients with melanoma with different disease stage and course, and modulating effects of high-dose interferon-α 2b treatment. J. Transl. Med. 2010, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, srep15179. [Google Scholar] [CrossRef] [Green Version]
- Leslie, C.; Bowyer, S.E.; White, A.; Grieu-Iacopetta, F.; Trevenen, M.; Iacopetta, B.; Amanuel, B.; Millward, M. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology 2015, 47, 557–563. [Google Scholar] [CrossRef]
- Baumgartner, J.; Wilson, C.; Palmer, B.; Richter, D.; Banerjee, A.; McCarter, M. Melanoma Induces Immunosuppression by Up-Regulating FOXP3+ Regulatory T Cells. J. Surg. Res. 2007, 141, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Budhu, S.; Hellmann, M.D.; Postow, M.A.; Senbabaoglu, Y.; Manne, S.; Gasmi, B.; Liu, C.; Zhong, H.; Li, Y.; et al. Non-conventional Inhibitory CD4+Foxp3−PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018, 33, 1017–1032.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [Green Version]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef] [Green Version]
- Simeone, E.; Gentilcore, G.; Giannarelli, D.; Grimaldi, A.M.; Caracò, C.; Curvietto, M.; Esposito, A.; Paone, M.; Palla, M.; Cavalcanti, E.; et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol. Immunother. 2014, 63, 675–683. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Umansky, V.; Sevko, A.; Gebhardt, C.; Utikal, J. Myeloid-derived suppressor cells in malignant melanoma. J. Dtsch. Dermatol. Ges. 2014, 12, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, S.M.; Shabaneh, T.B.; Zhang, P.; Martyanov, V.; Li, Z.; Malik, B.T.; Wood, T.A.; Boni, A.; Molodtsov, A.; Angeles, C.V.; et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017, 77, 1599–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.; Chen, J.J.; Yang, P. Pathophysiology of Tumor-Associated Macrophages. Adv. Clin. Chem. 2008, 45, 199–223. [Google Scholar] [CrossRef]
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N.A.R.N.M. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Donzelli, S.; Milano, E.; Pruszko, M.; Sacconi, A.; Masciarelli, S.; Iosue, I.; Melucci, E.; Gallo, E.; Terrenato, I.; Mottolese, M.; et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res. 2018, 20, 59. [Google Scholar] [CrossRef]
- Liu, H.; Yang, L.; Qi, M.; Zhang, J. NFAT1 enhances the effects of tumor-associated macrophages on promoting malignant melanoma growth and metastasis. Biosci. Rep. 2018, 38, 38. [Google Scholar] [CrossRef] [Green Version]
- Wanderley, C.W.; Colón, D.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; A Leite, C.; A Pereira, J.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [Green Version]
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.L.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Xiao, M.; Ge, Y.; Krepler, C.; Belser, E.; Coral, A.L.; Xu, X.; Zhang, G.; Azuma, R.; Liu, Q.; et al. BRAF Inhibition Stimulates Melanoma-Associated Macrophages to Drive Tumor Growth. Clin. Cancer Res. 2015, 21, 1652–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nat. Cell Biol. 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Kuklinski, L.F.; Yan, S.; Li, Z.; Fisher, J.L.; Cheng, C.; Noelle, R.J.; Angeles, C.V.; Turk, M.J.; Ernstoff, M.S. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol. Immunother. 2018, 67, 1113–1121. [Google Scholar] [CrossRef]
- Lines, J.L.; Sempere, L.F.; Wang, L.; Pantazi, E.; Mak, J.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Kakavand, H.; A Jackett, L.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676. [Google Scholar] [CrossRef]
- Rosenbaum, S.R.; Knecht, M.; Mollaee, M.; Zhong, Z.; Erkes, D.A.; McCue, P.A.; Chervoneva, I.; Berger, A.C.; Lo, J.A.; Fisher, D.E.; et al. FOXD3 Regulates VISTA Expression in Melanoma. Cell Rep. 2020, 30, 510–524.e6. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumor-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Klarquist, J.S.; Janssen, E.M. Melanoma-infiltrating dendritic cells. OncoImmunology 2012, 1, 1584–1593. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Domínguez, C.; Calderón-González, R.; Terán-Navarro, H.; Salcines-Cuevas, D.; García-Castaño, A.; Freire, J.; Gómez-Román, J.; Rivera, F. Dendritic cell therapy in melanoma. Ann. Transl. Med. 2017, 5, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Dakic, A. Development of dendritic cell system. Cell. Mol. Immunol. 2004, 1, 112–118. [Google Scholar] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- González, M.L.; Oosterhoff, D.; Lindenberg, J.J.; Milenova, I.; Lougheed, S.M.; Martiáñez, T.; Dekker, H.; Quixabeira, D.C.A.; Hangalapura, B.; Joore, J.; et al. Constitutively active GSK3β as a means to bolster dendritic cell functionality in the face of tumour-mediated immune suppression. OncoImmunology 2019, 8, e1631119-18. [Google Scholar] [CrossRef]
- Van De Ven, R.; Lindenberg, J.J.; Oosterhoff, D.; De Gruijl, T.D. Dendritic Cell Plasticity in Tumor-Conditioned Skin: CD14+ Cells at the Cross-Roads of Immune Activation and Suppression. Front. Immunol. 2013, 4, 403. [Google Scholar] [CrossRef] [Green Version]
- González, M.L.; Van De Ven, R.; De Haan, H.; Sluijs, J.V.E.V.D.; Dong, W.; Van Beusechem, V.W.; De Gruijl, T.D. Oncolytic adenovirus ORCA-010 increases the type 1 T cell stimulatory capacity of melanoma-conditioned dendritic cells. Clin. Exp. Immunol. 2020, 201, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Slone, N.; Chrisikos, T.T.; Kyrysyuk, O.; Babcock, R.L.; Medik, Y.B.; Li, H.S.; Kleinerman, E.S.; Watowich, S.S. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103+conventional dendritic cells. J. Immunother. Cancer 2020, 8, e000474. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-L.; Lee, Y.-P.; Pang, C.-Y.; Lin, H.-R.; Chen, C.-S.; Wen-Sheng, W. Tyrosine kinase inhibitors modulate dendritic cell activity via confining c-Kit signaling and tryptophan metabolism. Int. Immunopharmacol. 2020, 82, 106357. [Google Scholar] [CrossRef]
- Riegel, K.; Schlöder, J.; Sobczak, M.; Jonuleit, H.; Thiede, B.; Schild, H.; Rajalingam, K. RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell Death Differ. 2019, 27, 1300–1315. [Google Scholar] [CrossRef]
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and tumor progression of melanoma: New therapeutic prospectives. J. Immunotoxicol. 2012, 10, 235–252. [Google Scholar] [CrossRef]
- You, D.; Jung, S.P.; Jeong, Y.; Bae, S.Y.; Lee, J.E.; Kim, S. Fibronectin expression is upregulated by PI-3K/Akt activation in tamoxifen-resistant breast cancer cells. BMB Rep. 2017, 50, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorenko, I.V.; Abel, E.V.; Koomen, J.M.; Fang, B.; Wood, E.R.; Chen, Y.A.; Fisher, K.J.; Iyengar, S.; Dahlman, K.B.; Wargo, J.A.; et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 2015, 35, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Felding-Habermann, B.; Ruggeri, Z.M.; A Cheresh, D. Distinct biological consequences of integrin alpha v beta 3-mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. J. Biol. Chem. 1992, 267, 5070–5077. [Google Scholar]
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc. Natl. Acad. Sci. USA 2019, 116, 20141–20150. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix Metalloproteinases in Human Melanoma. J. Investig. Dermatol. 2000, 115, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; Vazquez, V.D.L.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870. https://doi.org/10.3390/cancers12102870
Falcone I, Conciatori F, Bazzichetto C, Ferretti G, Cognetti F, Ciuffreda L, Milella M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers. 2020; 12(10):2870. https://doi.org/10.3390/cancers12102870
Chicago/Turabian StyleFalcone, Italia, Fabiana Conciatori, Chiara Bazzichetto, Gianluigi Ferretti, Francesco Cognetti, Ludovica Ciuffreda, and Michele Milella. 2020. "Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy" Cancers 12, no. 10: 2870. https://doi.org/10.3390/cancers12102870
APA StyleFalcone, I., Conciatori, F., Bazzichetto, C., Ferretti, G., Cognetti, F., Ciuffreda, L., & Milella, M. (2020). Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers, 12(10), 2870. https://doi.org/10.3390/cancers12102870