From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
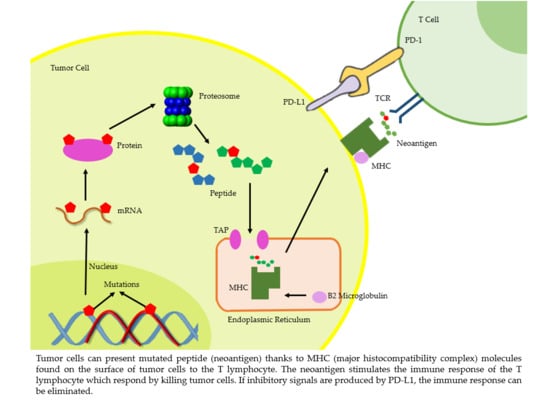
1. Introduction
2. TMB and Lung Cancer
2.1. What is TMB and How to Measure?
2.2. Clinical Features of TMB
2.3. Measurement of TMB in Tumor Tissue (tTMB)
2.4. Blood-Based Tumor Mutation Burden (bTMB)
3. TMB, From Great Expectation
3.1. Studies as Predictive Factor (Table)
3.2. Strengths
4. To Important Doubts
4.1. Limitations
4.2. Studies as Negative Predictive Factor
5. Is the TMB Dead?
6. New Emerging Biomarkers: TCRB T Cell Receptor Beta (TCRβ)
6.1. What is TCRB and How to Measure
6.2. Studies as Predictive Factor
6.3. Strengths
6.4. Limitations
7. Correlation TMB-TCRB
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2015, 8, 1–17. [Google Scholar] [CrossRef]
- Heeke, S.; Hofman, P. Tumor mutational burden assessment as a predictive biomarker for immunotherapy in lung cancer patients: Getting ready for prime-time or not? Transl. Lung Cancer Res. 2018, 7, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Nat. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [Green Version]
- Van Buuren, M.M.; Calis, J.J.; Schumacher, T.N. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. OncoImmunology 2014, 3, e28836. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Robbins, P.F.; Stroobant, V.; Vigneron, N.; Li, Y.F.; El-Gamil, M.; Hanada, K.-I.; Yang, J.C.; Rosenberg, S.A.; Eynde, B.J.V.D. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Nat. Acad. Sci. USA 2011, 108, e323–e331. [Google Scholar] [CrossRef] [Green Version]
- Linnemann, C.; Van Buuren, M.M.; Bies, L.; E Verdegaal, E.M.; Schotte, R.; A Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; El Atmioui, D.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Mcgranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Kumar Saini, S.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M.; Hirsch, F.R.; Hwang, D.; Botling, J.; Lopez-Rios, F.; Bubendorf, L.; Mino-Kenudson, M.; Roden, A.C.; Beasley, M.B.; Borczuk, A.; et al. The Promises and Challenges of Tumor Mutation Burden as an Immunotherapy Biomarker: A Perspective from the International Association for the Study of Lung Cancer Pathology Committee. J. Thorac. Oncol. 2020, 15. [Google Scholar] [CrossRef]
- Bodor, J.N.; Boumber, Y.; Borghaei, H. Biomarkers for immune checkpoint inhibition in non-small cell lung cancer (NSCLC). Cancer 2020, 126, 260–270. [Google Scholar] [CrossRef]
- Willis, C.; Fiander, M.; Tran, D.; Korytowsky, B.; Thomas, J.-M.; Calderon, F.; Zyczynski, T.M.; Brixner, D.; Stenehjem, D.D. Tumor mutational burden in lung cancer: A systematic literature review. Oncotarget 2019, 10, 6604–6622. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.F.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Diem, S.; Schmid, S.; Krapf, M.; Flatz, L.; Born, D.; Jochum, W.; Templeton, A.J.; Früh, M. Neutrophil-to-Lymphocyte ratio (NLR) and Platelet-to-Lymphocyte ratio (PLR) as prognostic markers in patients with non-small cell lung cancer (NSCLC) treated with nivolumab. Lung Cancer 2017, 111, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nat. Cell Biol. 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Galeas, J.; Cheng, H. Tumor mutation burden in lung cancer: A new predictive biomarker for immunotherapy or too soon to tell? J. Thorac. Dis. 2018, 10 (Suppl. S33), S3994–S3998. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- FDA Approves Second Biomarker Based Indication for Mercks KEYTRUDA-pembrolizumab-Regardless of Tumor Type. Available online: https://www.mrknewsroom.com/newsroom/news-releases/news-details/2020/FDA-Approves-Second-Biomarker-Based-Indication-for-Mercks-KEYTRUDA-pembrolizumab-Regardless-of-Tumor-Type/default.aspx (accessed on 17 June 2020).
- Xiao, D.; Pan, H.; Li, F.; Wu, K.; Zhang, X.; He, J. Analysis of ultra-deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget 2016, 7, 22857–22864. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Europe PMC Funders Group Mutational signatures associated with tobacco smoking in human cancer. Science 2018, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nat. Cell Biol. 2010, 461, 272–276. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nat. Cell Biol. 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Rudin, C.M.; Avila-Tang, E.; Harris, C.C.; Herman, J.G.; Hirsch, F.R.; Pao, W.; Schwartz, A.G.; Vahakangas, K.H.; Samet, J.M. Lung cancer in never smokers: Molecular profiles and therapeutic implications. Clin. Cancer Res. 2009, 15, 5646–5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.-M.; Lira, M.; Pandya, K.; Choi, Y.-L.; Ahn, J.S.; Mao, M.; Han, J.; Park, K.; Ahn, M.-J.; Kim, J. Clinical characteristics associated with ALK rearrangements in never-smokers with pulmonary adenocarcinoma. Lung Cancer 2014, 83, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Hendriks, L.; Cabrera, C.; Reguart, N.; Besse, B. Immunotherapy for oncogenic-driven advanced non-small cell lung cancers: Is the time ripe for a change? Cancer Treat. Rev. 2018, 71, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Berland, L.; Heeke, S.; Humbert, O.; Macocco, A.; Long-Mira, E.; Lassalle, S.; Lespinet-Fabre, V.; Lalvée, S.; Bordone, O.; Cohen, C.; et al. Current views on tumor mutational burden in patients with non-small cell lung cancer treated by immune checkpoint inhibitors. J. Thorac. Dis. 2019, 11 (Suppl. S1), S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, K.; Nagashima, T.; Urakami, K.; Ohshima, K.; Serizawa, M.; Ohnami, S.; Shimoda, Y.; Ohnami, S.; Maruyama, K.; Naruoka, A.; et al. Tumor mutational burden analysis of 2000 Japanese cancer genomes using whole exome and targeted gene panel sequencing. Biomed. Res. 2018, 39, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; Heuvel, M.M.V.D.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Med, C.C. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open- label, phase 1, multicohort study. Lancet Oncol. 2015, 40, 1291–1296. [Google Scholar]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Devarakonda, S.; Rotolo, F.; Tsao, M.-S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Velcheti, V.; Kim, E.S.; Mekhail, T.; Dakhil, C.; Stella, P.J.; Shen, X.; Hu, S.; Paul, S.M.; Shames, D.S.; Yun, C.; et al. Prospective clinical evaluation of blood-based tumor mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC): Interim B-F1RST results. J. Clin. Oncol. 2018, 36, 12001. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanderWalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J.; VanderWalde, A. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.; Gettinger, S.; Johnson, M.L.; Jänne, P.A.; Garassino, M.; Christoph, D.; Toh, C.K.; Rizvi, N.A.; Chaft, J.E.; Costa, E.C.; et al. Phase II Trial of Atezolizumab As First-Line or Subsequent Therapy for Patients With Programmed Death-Ligand 1–Selected Advanced Non-Small-Cell Lung Cancer (BIRCH). J. Clin. Oncol. 2017, 35, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Evaluation of Automatic Class III Designation for MSK—IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets). Available online: https://www.accessdata.fda.gov/cdrh_docs/reviews/DEN170058 (accessed on 15 November 2017).
- Oncomine TM Tumor Mutation Load Assay. Available online: http://assets.thermofisher.com/TFS-Assets/CSD/Flyers/oncomine-tumor-mutation-load-assay-flyer.pdf (accessed on 4 October 2018).
- Kowanetz, M.; Zou, W.; Shames, D.; Cummings, C.; Rizvi, N.; Spira, A.; Frampton, G.; Leveque, V.; Flynn, S.; Mocci, S.; et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 2016, 27 (Suppl. S6), vi23. [Google Scholar] [CrossRef]
- Teraoka, S.; Fujimoto, D.; Morimoto, T.; Kawachi, H.; Ito, M.; Sato, Y.; Nagata, K.; Nakagawa, A.; Otsuka, K.; Uehara, K.; et al. Early Immune-Related Adverse Events and Association with Outcome in Advanced Non-Small Cell Lung Cancer Patients Treated with Nivolumab: A Prospective Cohort Study. J. Thorac. Oncol. 2017, 12, 1798–1805. [Google Scholar] [CrossRef]
- Judd, J.; Zibelman, M.; Handorf, E.; O’Neill, J.; Ramamurthy, C.; Bentota, S.; Doyle, J.; Uzzo, R.G.; Bauman, J.; Borghaei, H.; et al. Immune-Related Adverse Events as a Biomarker in Non-Melanoma Patients Treated with Programmed Cell Death 1 Inhibitors. Oncology 2017, 22, 1232–1237. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, Y.; Yoshino, K.; Otsuka, A.; Funakoshi, T.; Uchi, H.; Fujimura, T.; Matsushita, S.; Hata, H.; Okuhira, H.; Tanaka, R.; et al. Retrospective study of advanced melanoma patients treated with ipilimumab after nivolumab: Analysis of 60 Japanese patients. J. Dermatol. Sci. 2018, 89, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.I.; Kim, M.; Lee, S.-H.; Park, S.Y.; Kim, Y.N.; Kim, H.; Jeon, M.J.; Kim, T.Y.; Kim, S.W.; Kim, W.B.; et al. Development of thyroid dysfunction is associated with clinical response to PD-1 blockade treatment in patients with advanced non-small cell lung cancer. OncoImmunology 2018, 7, e1375642. [Google Scholar] [CrossRef] [Green Version]
- Rzepecki, A.K.; Cheng, H.; McLellan, B.N. Cutaneous toxicity as a predictive biomarker for clinical outcome in patients receiving anticancer therapy. J. Am. Acad. Dermatol. 2018, 79, 545–555. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Loewer, M.; Van De Roemer, N.; De Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bate, A.; Evans, S.J.W. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol. Drug Saf. 2009, 18, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Geffen, J.; Forster, K. Treatment of ADHD in adults. Ther. Adv. Vaccin. 2018, 8, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Herbst, R.; Lopes, G.; Kowalski, D.; Nishio, M.; Wu, Y.-L.; Junior, G.D.C.; Baas, P.; Kim, D.-W.; Gubens, M.; Cristescu, R.; et al. Association between tissue TMB (tTMB) and clinical outcomes with pembrolizumab monotherapy (pembro) in PD-L1-positive advanced NSCLC in the KEYNOTE-010 and -042 trials. Ann. Oncol. 2019, 30, v916–v917. [Google Scholar] [CrossRef]
- tTMB is Not Established as a Marker for Pembrolizumab Efficacy in NSCLC. Two Exploratory Analysis of the Relationship Between Tissue Tumour Mutational Burden and Pembrolizumab Efficacy Provided Conflicting Findings. Available online: https://www.esmo.org/oncology-news/tTMB-Is-Not-Established-as-a-aMarker-for-Pembrolizumab-Efficacy-in-NSCLC. (accessed on 27 September 2019).
- Garassino, M.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Cristescu, R.; et al. OA04.06 Evaluation of TMB in KEYNOTE-189: Pembrolizumab Plus Chemotherapy vs Placebo Plus Chemotherapy for Nonsquamous NSCLC. J. Thorac. Oncol. 2019, 14, S216–S217. [Google Scholar] [CrossRef]
- Langer, C.; Gadgeel, S.; Borghaei, H.; Patnaik, A.; Powell, S.; Gentzler, R.; Yang, J.; Gubens, M.; Sequist, L.; Awad, M.; et al. OA04.05 KEYNOTE-021: TMB and Outcomes for Carboplatin and Pemetrexed With or Without Pembrolizumab for Nonsquamous NSCLC. J. Thorac. Oncol. 2019, 14, S216. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Langer, C.; Novello, S.; Halmos, B.; Cheng, Y.; Gadgeel, S.; Hui, R.; Sugawara, S.; Borghaei, H.; Cristescu, R.; et al. Pembrolizumab (pembro) plus platinum-based chemotherapy (chemo) for metastatic NSCLC: Tissue TMB (tTMB) and outcomes in KEYNOTE-021, 189, and 407. Ann. Oncol. 2019, 30 (Suppl. S5), v917–v918. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Costa, E.C.; Park, K.; Alexandru, A.; Lupinacci, L.; Jimenez, E.D.L.M.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Nat. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Lisberg, A.; Zaretsky, J.M.; Grogan, T.R.; Rizvi, H.; Wells, D.K.; Carroll, J.; Cummings, A.; Madrigal, J.; Jones, B.; et al. Tumor Characteristics Associated with Benefit from Pembrolizumab in Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 5061–5068. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2019, 20892, 1–5. [Google Scholar] [CrossRef]
- Looney, T.J.; Topacio-Hall, D.; Lowman, G.; Conroy, J.; Morrison, C.; Oh, D.; Fong, L.; Zhang, L. TCR Convergence in Individuals Treated With Immune Checkpoint Inhibition for Cancer. bioRxiv 2019, 665612. [Google Scholar] [CrossRef]
- Zhang, L.; Looney, T.; Topacio-Hall, D.; Lowman, G.; Oh, D.; Fong, L. Peripheral Blood TCRB Repertoire Convergence and Clonal Expansion Predict Response to Anti-CTLA-4 Monotherapy for Cancer; Thermo Fisher Scientific: Waltham, MA, USA, 2018. [Google Scholar]
- Leonards, K.; Alborelli, I.; Looney, T.; Quagliata, L.; Jermann, P.M. Elevated TCRB repertoire convergence and clonal expansion in the NSCLC tumour microenvironment of responders to anti-PD-1 monotherapy. Ecp2019 2019, 1, 7. [Google Scholar]
- McNeel, D.G. TCR diversity—A universal cancer immunotherapy biomarker? J. Immunother. Cancer 2016, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Cha, E.; Klinger, M.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved Survival with T Cell Clonotype Stability After Anti-CTLA-4 Treatment in Cancer Patients. Sci. Transl. Med. 2014, 6, 238ra70. [Google Scholar] [CrossRef] [Green Version]
- Storkus, W.J.; Lin, Y.; Miller, L.; Topacio-Hall, D.S.; Lowman, G.M.; Looney, T.; Butterfield, L.H.; Taylor, J.L.; Tarhini, A.A.; Tawbi, H.A.; et al. Peripheral Blood TCRB Chain Convergence Predicts Response to Dendritic Cell- Based Immunotherapy in Advanced-Stage Melanoma Patients. J. Immunother. Cancer 2018, 6, 12060479. [Google Scholar] [CrossRef]
- Looney, T.; Lowman, G.; Miller, L.; Linch, E. TCR beta chain convergence defines the tumor infiltrating T cell repertoire of melanoma and non-small cell lung carcinoma. Ann. Oncol. 2018, 29 (Suppl. S8), viii19–viii20. [Google Scholar] [CrossRef]
- Looney, T.; Miller, L.; Lowman, G.; Linch, E.; Zheng, J.; Topacio-Hall, D. Evidence for antigen-driven TCRB chain convergence in the tumourinfiltrating t cell repertoire of 148 research subjects with melanoma. ESMO Open 2018, 3, A10.2–A11. [Google Scholar]
- Liu, Y.; Yang, Q.; Yang, J.; Cao, R.; Liang, J.; Liu, Y.; Zeng, Y.; Chen, S.; Xia, X.; Zhang, K.; et al. Characteristics and prognostic significance of profiling the peripheral blood T-cell receptor repertoire in patients with advanced lung cancer. Int. J. Cancer 2019, 145, 1423–1431. [Google Scholar] [CrossRef]
- Song, L.; He, Y.; Li, X.; Zhou, C. 1248PAlterations of TMB and TCR repertoires during chemotherapy in East Asian lung cancer patients without TKI-related driver gene mutations. Ann. Oncol. 2019, 30 (Suppl. S5), v509. [Google Scholar] [CrossRef]
- Wang, F.; Xie, X.; Song, M.; Ji, L.; Liu, M.; Li, P.; Guan, Y.; Lin, X.; Qin, Y.; Xie, Z.; et al. Tumor immune microenvironment and mutational analysis of tracheal adenoid cystic carcinoma. Ann. Transl. Med. 2020, 8, 750. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |

{kind=link}
| Drug Trial | Study Type/Phase | Line of Therapy | Pts, n | Patient Population | Tmb Method & Cutoff | Clinical Outcomes | Author/Year |
|---|---|---|---|---|---|---|---|
| Pembrolizumab | Retrospective | First line, second or higher | 16 of POPLAR trial; 18 of OAK study | Advanced NSCLC | WES: high≥ 178 mutations | TMB was correlated with better ORR (63% vs. 0%, p = 0.03), PFS (14.5 vs. 3.7 m, p = 0.01) and DCB. | Rizvi NA 2015 [31] |
| CHECKMATE-026 Nivolumab (NCT02041533) | Exploratory retrospective analysis of phase III study | First line | 312 | Stage IV or recurrent NSCLC with PD-L1 ≥1% | WES: highTMB ≥243; low TMB <100 mutations | High TMB pts: PFS 9.7 vs. 5.8 m (HR 0.62; 95% CI, 0.38 to 1.00) and ORR (46.8% vs. 28.3%) in nivolumab group compared to chemotherapy. | Carbone D,2017 [33] |
| CHECKMATE-012 Nivolumab& ipilimumab (NCT01454102) | Phase I | First line | 75 | Advanced NSCLC | WES: high TMB > median, 158 mutations; low TMB ≤ median | ORR, DCB, PFS were superior in pts with high TMB vs. low TMB (ORR 51% vs. 13%, p = 0.0005; DCB 65% vs. 34%, p = 0.011; PFS HR 0.41). | Hellmann MD 2018 [34] |
| CHECKMATE-227 Nivolumab + ipilimumab (NCT02477826) | Phase III | First line | 299 | Stage IV or recurrent NSCLC | FoundationOne CDx assay; high TMB: ≥10 mut/MbV | PFS was longer among pts with high TMB (mPFS: 7.2 vs. 5.5 months, HR 0.58, p < 0.001) in nivolumab + ipilimumab group compared to chemotherapy | Hellman MD 2018 [35] |
| CHECKMATE-568 Nivolumab + ipilimumab (NCT02659059) | Phase II | First line | 288 | Stage IV NSCLC | FoundationOne CDx assay; high TMB: ≥10 mut/Mb | ORR was higher (>40%) in high TMB | Ramalingam SS 2018 [32] |
| CHECKMATE-032 Nivolumab ± ipilimumab (NCT01928394) | Exploratory | Second-line or higher | 211 | Advanced SCLC | WES: TMB was grouped by tertiles: low, 0 to <143; medium, 143 to 247; high, ≥248 mutations | ORR: 46.2% vs.16%; 1-year PFS: 30% vs. 6.2% 1-year OS: 62.4% vs. 23.4% was higher in pts with TMB high vs TMB low | Hellmann MD 2018 [36] |
| PD-1 or PD-L1 inhibitors | Retrospective | First line, second or higher | 240 | Advanced NSCLC | MSK-IMPACT TMB was grouped by percentiles: high TMB >50% | More disease control (complete/partial response vs stable/progression disease) and longer PFS for patients with high TMB >50% | Rizvi H, 2018 [25] |
| POPLAR & OAK Atezolizumab | Retrospective | Second-line or higher | 211 (discovery cohort with 16 p) in POPLAR trial, 583 (validating cohort with 18 p) in OAK study | Advanced NSCLC | Foundation One; bTMB: High bTMB ≥16; low TMB ≤16. | High bTMB (≥16 mut/Mb) was associated with improved PFS, ORR and duration of response. | Gandara DR, 2018 [37] |
| LACE-BIO-2 Adjuvant Cisplatin (NCT01294280) | Retrospective | Adjuvant chemotherapy | >900 | Early-stage NSCLC | Targeted NGS panel using Illumina HiSeq 2000. TMB was categorized into tertiles (low, ≤4 mutations/Mb; intermediate, >4 and ≤8 mutations/Mb; high, >8 mutations/Mb) | High TMB (>8 mut/Mb) was prognostic for favorable OS, PFS, LCSS in patients with resected NSCLC. LCSS benefit with adjuvant chemotherapy was more pronounced in low TMBs (≤4 mut/Mb). | Devarakonda S, 2018 [38] |
| Neoadjuvant nivolumab | Exploratory | Neoadjuvant PD-1 Blockade | 22 (21 were eligible for inclusion) | Surgically resectable early (stage I, II, or IIIA) NSCLC. | WES: highTMB: 311 ± 55 media vs low TMB:74 ± 60 mean | In pts with high TMB (sequence alterations; mean, 311 ± 55 vs. 74 ± 60, p = 0.01) a major pathological response was observed. | Forde PM, 2018 [39] |
| B-F1RST Atezolizumab (NCT02848651) | Phase II | First line | 152 (119 were included in the biomarker evaluable population) | Locally advanced or metastatic NSCLC | Foundation Medicine panel; bTMB: high bTMB ≥ 16, versus low bTMB ≤ 16 | It was observed a relationship between increasing bTMB score and improved clinical outcomes. ORR and PFS were superior in pts with high bTMB vs low bTMB: ORR 28.6% vs. 4.4%; PFS 4.6 months vs. 3.7 months, HR 0.66 (90% CI 0.42–1.02). | Velcheti V, 2018 [40] |
| Drug Trial | Study Type | Pts, n | Patient Population | Purpose of Study | Tmb Method & Cutoff | Clinical Outcomes | Conclusion |
|---|---|---|---|---|---|---|---|
| KEYNOTE-010 (NCT01905657) | Exploratory retrospective analysis of a randomised controlled trial phase II/III | 253 (24% from the all sample) | Previously treated or untreated advanced NSCLC PD-L1(+) with tumour proportion score (TPS)≥ 1% having evaluable Ttmb | Association between tTMB and clinical benefit with pembrolizumab monotherapy | tTMB determined by WES of tumour and matched normal DNA Cutpoint of 175 mutations per exome | tTMB ≥ 175: OS 14.1 m vs. 7.6 m (CI, 0.38–0.83); PFS 4.2 m vs. 2.4 m (CI, 0.40–0.87); ORR 23.5% vs. 9.8% with pembrolizumab and chemotherapy respectively | tTMB was associated with OS, PFS and ORR for the pembrolizumab arms but tTMB was not associated with outcomes for chemotherapy [59,60]. |
| KEYNOTE-042 (NCT02220894) | Exploratory retrospective analysis of a randomised controlled trial phase III | 793 (62% from the all sample) | tTMB ≥ 175: OS 21.9 m vs. 11.6 m (CI, 0.48–0.80); PFS 6.3 m vs. 6.5 m (CI, 0.59–0.95); ORR 34.4% vs. 30.9% with pembrolizumab and chemotherapy respectively | ||||
| KEYNOTE-021 (NCT02039674) | Exploratory analysis of a randomised controlled trial phase I/II study | 267 (48% of patients in cohorts C and G) | Stage IIIb/IV non-squamous NSCLC | Association of tTMB with outcomes for pembrolizumab + chemotherapy and for chemotherapy | tTMB determined by WES of tumour and matched normal DNA Cutpoint of 175 mutations per exome | In cohort G, ORR was higher with pembrolizumab + chemotherapy vs chemotherapy in the 31 patients with tTMB ≥175 mutations per exome (71.4% vs. 30%) | No significant association was determined between tTMB and efficacy of pembrolizumab + chemotherapy or chemotherapy alone. TMB does not seem to identify responders from non responders either for the combination treatment or chemotherapy alone. [60,61,62,63]. |
| KEYNOTE-189 (NCT02578680) | Exploratory analysis of a randomised controlled trial phase III | 616 (48% from the all sample) | tTMB ≥ 175: OS was improved with pembrolizumab + chemotherapy over chemotherapy (HR 0.64; CI 0.38-1.07), PFS (HR 0.32; CI 0.21-0´51) | ||||
| KEYNOTE-407 (NCT02775435) | Exploratory analysis of a randomised controlled phase III study | 559 (56% from the all sample) | Stage IV squamous NSCLC | tTMB ≥ 175: OS was improved with pembrolizumab + chemotherapy over chemotherapy (HR 0.74; CI 0.50-1.08), PFS (HR 0.57; CI 0.41-0.81) | |||
| CHECKMATE-026 (NCT02041533) | Exploratory analysis of randomised phase III study | 312 (58% of the patients who had undergone randomization) | Stage IV or recurrent (PD-L1)–positive NSCLC | Assess the effect of the TMB on outcomes with nivolumab vs. docetaxel | TMB determined in tumor and blood samples by WES 0to100 (low burden) 100to242 (medium) ≥ 243 (high) | tTMB ≥ 243: ORR was higher in the nivolumab group than in the chemotherapy (47% vs. 28%), and PFS was longer (median, 9.7 vs. 5.8 months; HR 0.62; 95% CI, 0.38 to 1.00). | No significant difference was observed in OS between the nivolumab and chemotherapy groups regardless of TMB, according to findings published in the New England Journal of Medicine [33]. |
| CHECKMATE-227 (NCT02477826) | Exploratory analysis of randomised phase III study | 679 (58.2% from the all sample) | Stage IV or recurrent NSCLC | Evaluate TMB as a potential predictive biomarker of efficacy of nivolumab, nivolumab + ipilimumab, nivolumab + platinum-doublet chemotherapy and of platinum-doublet. | TMB determined by WES Cutpoint of 10 mutations per megabase | Similar degree of OS benefit in nivolumab + ipilimumab, regardless of TMB (≥10 vs. <10 mutations per megabase, respectively) OS benefit for nivolumab plus ipilimumab vs chemotherapy regardless of TMB or PD-L1 | A similar degree of OS benefit was found for nivolumab and ipilimumab regardless of TMB according to findings published in the New England Journal of Medicine Combination of PD-L1 and TMB did not reveal a subgroup with an increased benefit for nivo + ipi vs chemotherapy [64]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sesma, A.; Pardo, J.; Cruellas, M.; Gálvez, E.M.; Gascón, M.; Isla, D.; Martínez-Lostao, L.; Ocáriz, M.; Paño, J.R.; Quílez, E.; et al. From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy. Cancers 2020, 12, 2974. https://doi.org/10.3390/cancers12102974
Sesma A, Pardo J, Cruellas M, Gálvez EM, Gascón M, Isla D, Martínez-Lostao L, Ocáriz M, Paño JR, Quílez E, et al. From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy. Cancers. 2020; 12(10):2974. https://doi.org/10.3390/cancers12102974
Chicago/Turabian StyleSesma, Andrea, Julián Pardo, Mara Cruellas, Eva M. Gálvez, Marta Gascón, Dolores Isla, Luis Martínez-Lostao, Maitane Ocáriz, José Ramón Paño, Elisa Quílez, and et al. 2020. "From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy" Cancers 12, no. 10: 2974. https://doi.org/10.3390/cancers12102974
APA StyleSesma, A., Pardo, J., Cruellas, M., Gálvez, E. M., Gascón, M., Isla, D., Martínez-Lostao, L., Ocáriz, M., Paño, J. R., Quílez, E., Ramírez, A., Torres-Ramón, I., Yubero, A., Zapata, M., & Lastra, R. (2020). From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy. Cancers, 12(10), 2974. https://doi.org/10.3390/cancers12102974