Implementing Systems Modelling and Molecular Imaging to Predict the Efficacy of BCL-2 Inhibition in Colorectal Cancer Patient-Derived Xenograft Models

 ,
,  , , , ,
, , , ,  ,
,  ,
,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
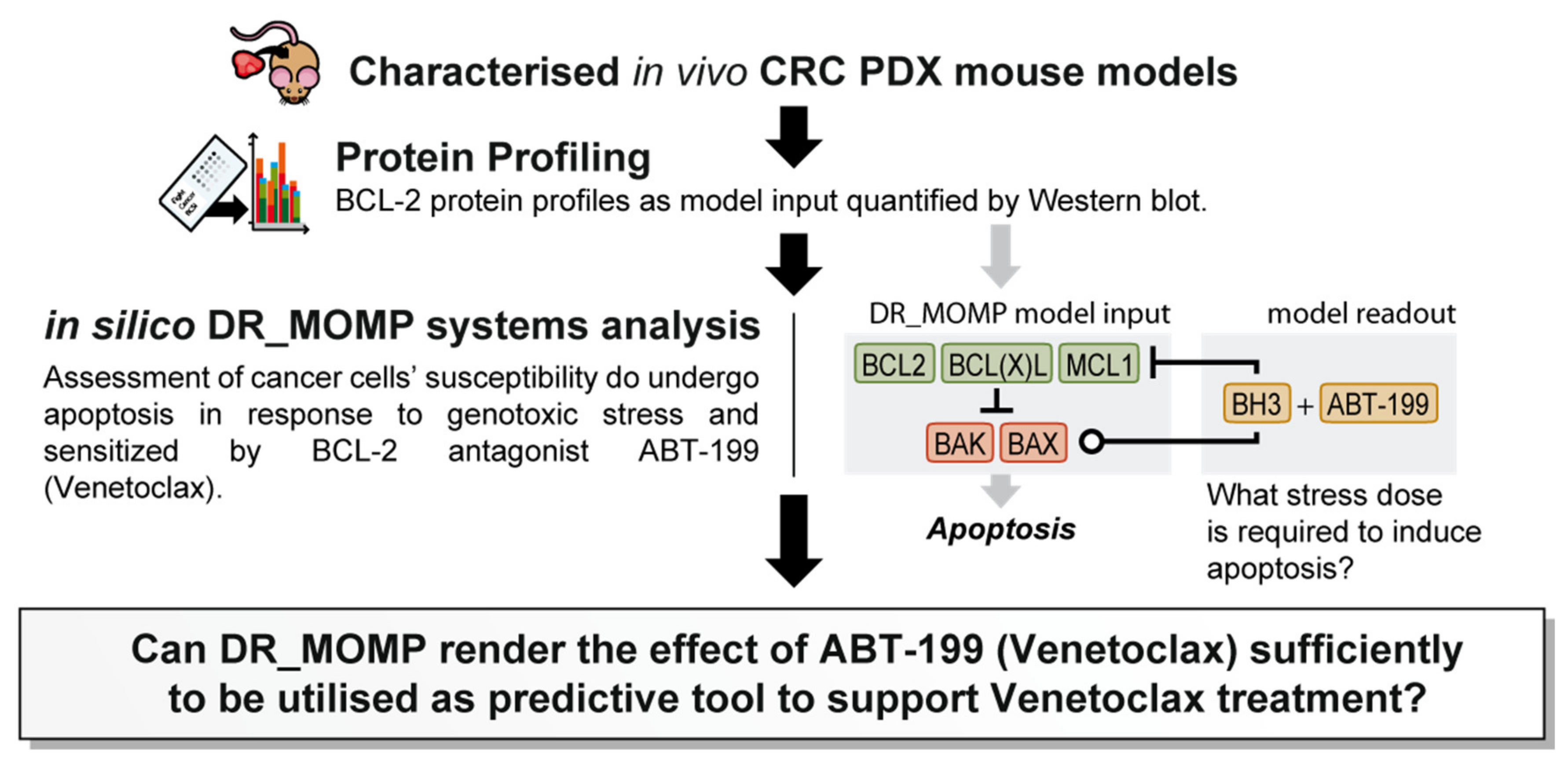
1. Introduction
2. Results
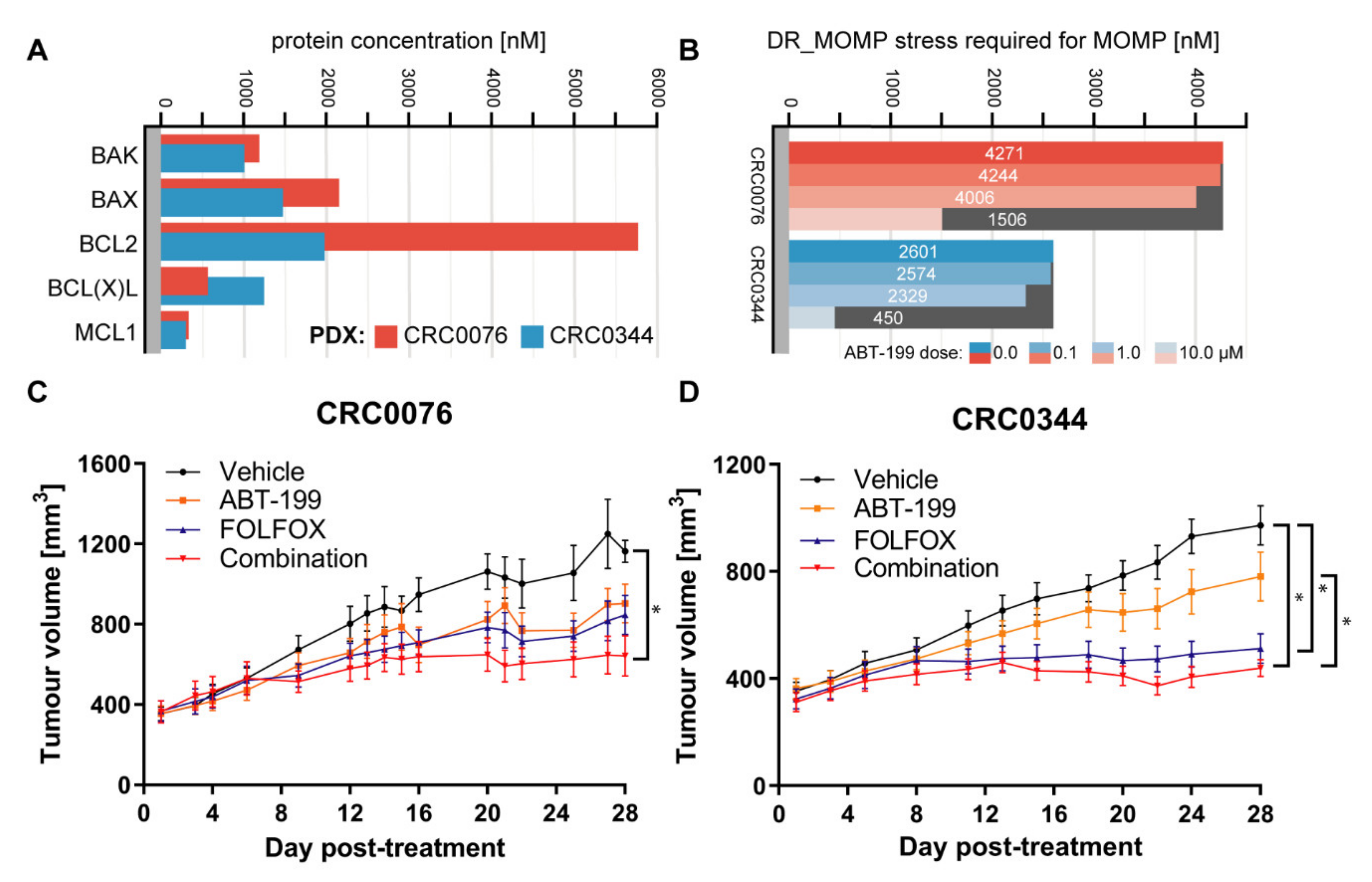
2.1. In Vivo PDX Response to ABT-199 Combined with FOLFOX Validates DR_MOMP Predictions
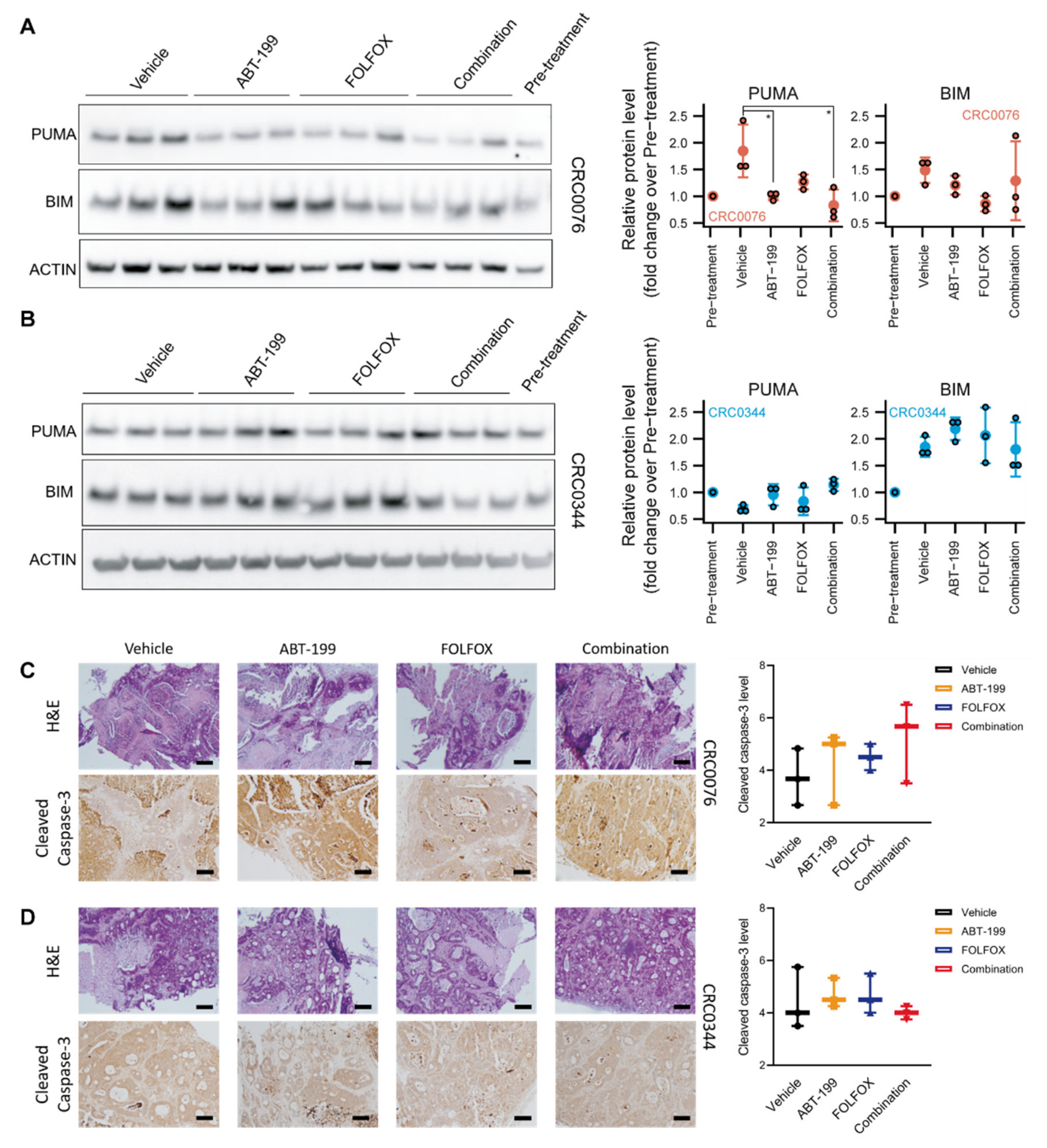
2.2. ABT-199 Treatment In Vivo Leads to Decrease of p53 Upregulated Modulator of Apoptosis (PUMA) Levels in the CRC0076 PDX Model
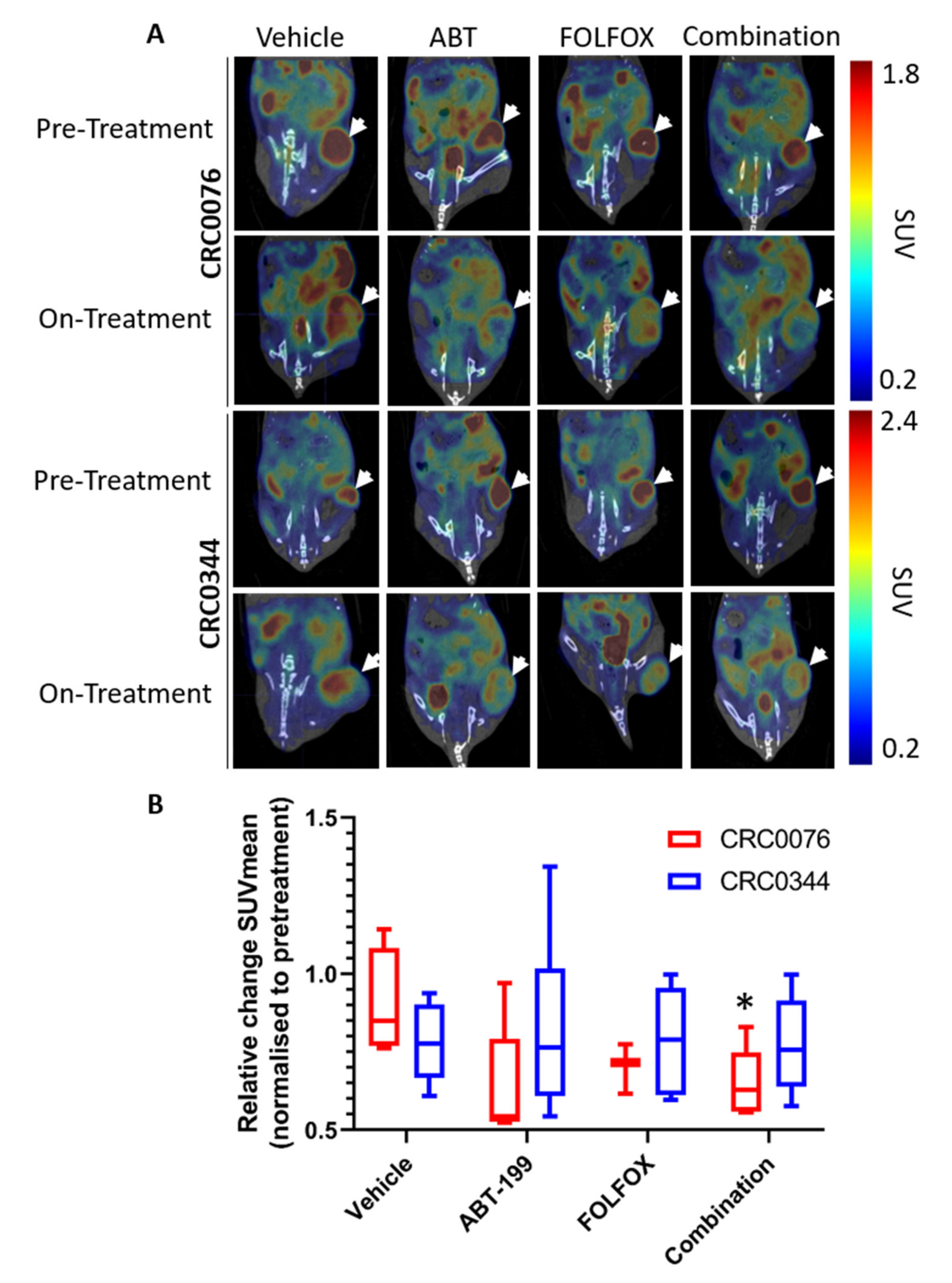
2.3. Decreased Tumour Glucose Uptake as an Early Biomarker of Response to FOLFOX and ABT-199 Combination Therapy
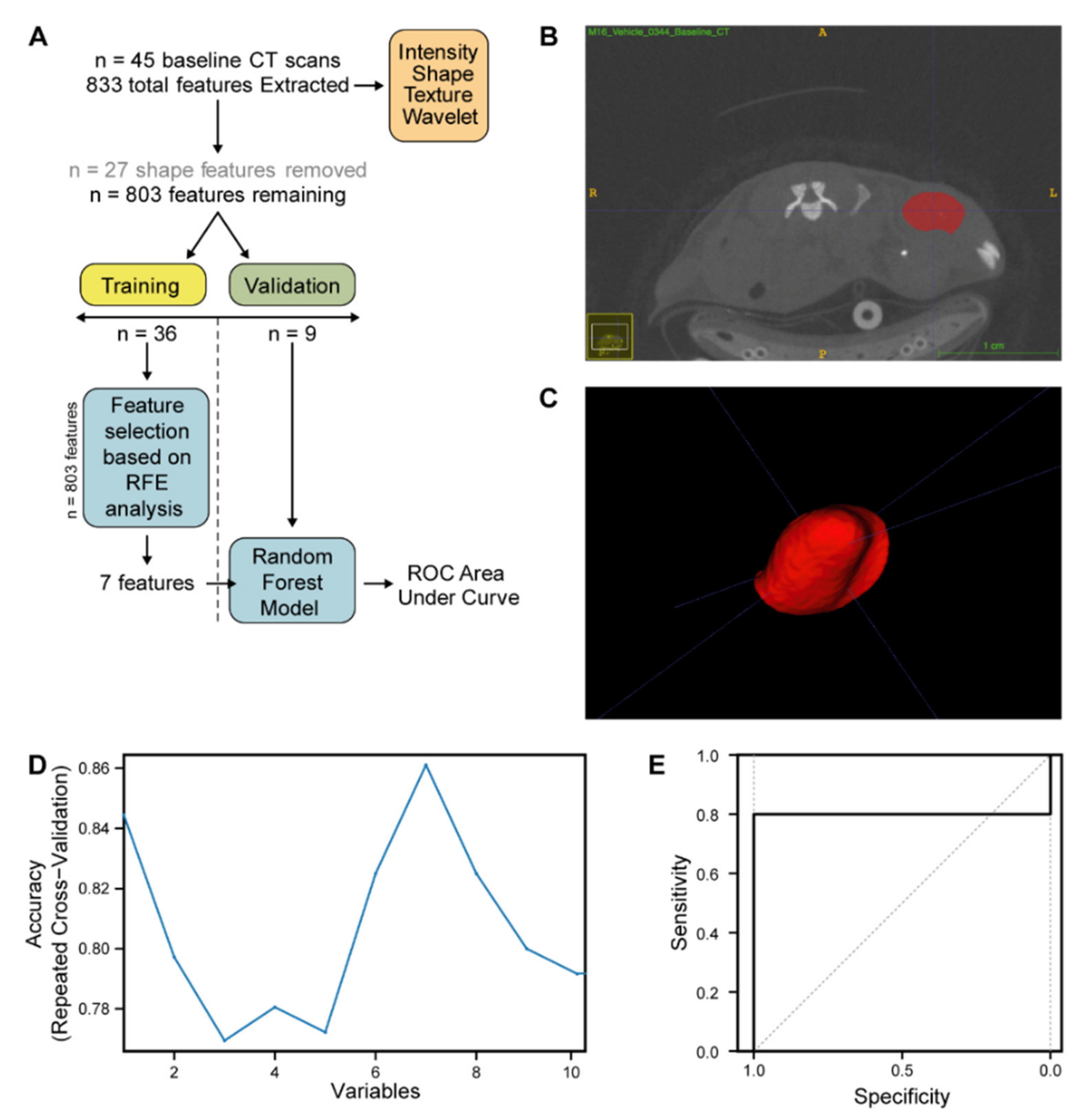
2.4. Radiomic CT Feature Analysis in DR_MOMP Predicted Combination-Only Responder (CRC0076) and FOLFOX Alone Responder (CRC0344) PDX Models
3. Discussion
4. Materials and Methods
4.1. Quantitative Western Blotting
4.2. DR_MOMP Calculation
4.3. Chemicals
4.4. Cell Lines
4.5. In Vitro Toxicity Assays and Synergy Calculations
4.6. Animals
4.7. PDX Efficacy Study
4.8. Immunohistochemistry (IHC)
4.9. 18F-FDG-PET/CT Study
4.10. Radiomic Analysis of CRC PDX CT Images
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Prescribing Information Reference ID 4275193, NDA 208573/S-004 and S-005. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/208573Orig1s004s005ltr.pdf (accessed on 1 July 2019).
- Vogler, M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv. Med. 2014, 2014, 943648. [Google Scholar] [CrossRef] [Green Version]
- Lindner, A.U.; Concannon, C.G.; Boukes, G.J.; Cannon, M.D.; Llambi, F.; Ryan, D.; Boland, K.; Kehoe, J.; McNamara, D.A.; Murray, F.; et al. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013, 73, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, L.; Lindner, A.U.; de Chaumont, C.; Kehoe, J.; Fay, J.; Bacon, O.; Toomey, S.; Huber, H.J.; Hennessy, B.T.; Kay, E.W.; et al. BCL2 protein signalling determines acute responses to neoadjuvant chemoradiotherapy in rectal cancer. J. Mol. Med. Berl. 2015, 93, 315–326. [Google Scholar] [CrossRef]
- Lindner, A.U.; Salvucci, M.; Morgan, C.; Monsefi, N.; Resler, A.J.; Cremona, M.; Curry, S.; Toomey, S.; O’Byrne, R.; Bacon, O.; et al. BCL-2 system analysis identifies high-risk colorectal cancer patients. Gut 2017, 66, 2141–2148. [Google Scholar] [CrossRef]
- Lucantoni, F.; Lindner, A.U.; O’Donovan, N.; Dussmann, H.; Prehn, J.H.M. Systems modeling accurately predicts responses to genotoxic agents and their synergism with BCL-2 inhibitors in triple negative breast cancer cells. Cell Death Dis. 2018, 9, 42. [Google Scholar] [CrossRef]
- Weiss, J.; Gajek, T.; Kohler, B.C.; Haefeli, W.E. Venetoclax (ABT-199) Might Act as a Perpetrator in Pharmacokinetic Drug-Drug Interactions. Pharmaceutics 2016, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Zanella, E.R.; Galimi, F.; Sassi, F.; Migliardi, G.; Cottino, F.; Leto, S.M.; Lupo, B.; Erriquez, J.; Isella, C.; Comoglio, P.M.; et al. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci. Transl. Med. 2015, 7, 272ra12. [Google Scholar] [CrossRef]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.; Pervez, S. Allred scoring for ER reporting and it’s impact in clearly distinguishing ER negative from ER positive breast cancers. J. Pak. Med. Assoc. 2010, 60, 350–353. [Google Scholar]
- Salvucci, M.; Wurstle, M.L.; Morgan, C.; Curry, S.; Cremona, M.; Lindner, A.U.; Bacon, O.; Resler, A.J.; Murphy, A.C.; O’Byrne, R.; et al. A Stepwise Integrated Approach to Personalized Risk Predictions in Stage III Colorectal Cancer. Clin. Cancer Res. 2017, 23, 1200–1212. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef]
- Hector, S.; Prehn, J.H. Apoptosis signaling proteins as prognostic biomarkers in colorectal cancer: A review. Biochim. Biophys. Acta 2009, 1795, 117–129. [Google Scholar] [CrossRef]
- Violette, S.; Poulain, L.; Dussaulx, E.; Pepin, D.; Faussat, A.M.; Chambaz, J.; Lacorte, J.M.; Staedel, C.; Lesuffleur, T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. Int. J. Cancer 2002, 98, 498–504. [Google Scholar] [CrossRef]
- Koehler, B.C.; Scherr, A.L.; Lorenz, S.; Urbanik, T.; Kautz, N.; Elssner, C.; Welte, S.; Bermejo, J.L.; Jager, D.; Schulze-Bergkamen, H. Beyond cell death—antiapoptotic Bcl-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS ONE 2013, 8, e76446. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Fricker, M.; O’Prey, J.; Tolkovsky, A.M.; Ryan, K.M. Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis. 2010, 1, e59. [Google Scholar] [CrossRef] [Green Version]
- Meller, R.; Cameron, J.A.; Torrey, D.J.; Clayton, C.E.; Ordonez, A.N.; Henshall, D.C.; Minami, M.; Schindler, C.K.; Saugstad, J.A.; Simon, R.P. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J. Biol. Chem. 2006, 281, 7429–7436. [Google Scholar] [CrossRef] [Green Version]
- Rehm, M.; Dussmann, H.; Janicke, R.U.; Tavare, J.M.; Kogel, D.; Prehn, J.H. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem. 2002, 277, 24506–24514. [Google Scholar] [CrossRef] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2-Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef]
- Boland, K.; Flanagan, L.; Prehn, J.H. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 2013, 4, e725. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Williams, A.; Hayashi, T.; Wolozny, D.; Yin, B.; Su, T.C.; Betenbaugh, M.J.; Su, T.P. The non-apoptotic action of Bcl-xL: regulating Ca(2+) signaling and bioenergetics at the ER-mitochondrion interface. J. Bioenerg. Biomembr. 2016, 48, 211–225. [Google Scholar] [CrossRef]
- Gross, A. BCL-2 family proteins as regulators of mitochondria metabolism. Biochim. Biophys. Acta 2016, 1857, 1243–1246. [Google Scholar] [CrossRef]
- Lucantoni, F.; Dussmann, H.; Llorente-Folch, I.; Prehn, J.H.M. BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 2018, 9, 26046–26063. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef]
- Rogers, W.; Thulasi Seetha, S.; Refaee, T.A.G.; Lieverse, R.I.Y.; Granzier, R.W.Y.; Ibrahim, A.; Keek, S.A.; Sanduleanu, S.; Primakov, S.P.; Beuque, M.P.L.; et al. Radiomics: from qualitative to quantitative imaging. Br. J. Radiol. 2020, 93, 20190948. [Google Scholar] [CrossRef]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images Are More than Pictures, They Are Data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; van Stiphout, R.G.; Granton, P.; Zegers, C.M.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Sanduleanu, S.; Woodruff, H.C.; de Jong, E.E.C.; van Timmeren, J.E.; Jochems, A.; Dubois, L.; Lambin, P. Tracking tumor biology with radiomics: A systematic review utilizing a radiomics quality score. Radiother. Oncol. 2018, 127, 349–360. [Google Scholar] [CrossRef]
- Chaddad, A.; Daniel, P.; Niazi, T. Radiomics Evaluation of Histological Heterogeneity Using Multiscale Textures Derived From 3D Wavelet Transformation of Multispectral Images. Front. Oncol. 2018, 8, 96. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alferez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinska, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Wu, G.; Woodruff, H.C.; Sanduleanu, S.; Refaee, T.; Jochems, A.; Leijenaar, R.; Gietema, H.; Shen, J.; Wang, R.; Xiong, J.; et al. Preoperative CT-based radiomics combined with intraoperative frozen section is predictive of invasive adenocarcinoma in pulmonary nodules: a multicenter study. Eur. Radiol. 2020, 30, 2680–2691. [Google Scholar] [CrossRef] [Green Version]
- Panth, K.M.; Leijenaar, R.T.; Carvalho, S.; Lieuwes, N.G.; Yaromina, A.; Dubois, L.; Lambin, P. Is there a causal relationship between genetic changes and radiomics-based image features? An in vivo preclinical experiment with doxycycline inducible GADD34 tumor cells. Radiother. Oncol. 2015, 116, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Lambin, P.; Leijenaar, R.T.H.; Deist, T.M.; Peerlings, J.; de Jong, E.E.C.; van Timmeren, J.; Sanduleanu, S.; Larue, R.; Even, A.J.G.; Jochems, A.; et al. Radiomics: the bridge between medical imaging and personalized medicine. Nat. Rev. Clin. Oncol. 2017, 14, 749–762. [Google Scholar] [CrossRef]
- Zinn, P.O.; Singh, S.K.; Kotrotsou, A.; Hassan, I.; Thomas, G.; Luedi, M.M.; Elakkad, A.; Elshafeey, N.; Idris, T.; Mosley, J.; et al. A Coclinical Radiogenomic Validation Study: Conserved Magnetic Resonance Radiomic Appearance of Periostin-Expressing Glioblastoma in Patients and Xenograft Models. Clin. Cancer Res. 2018, 24, 6288–6299. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.T.; Sinai, P.; Kain, S.R. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Anal. Biochem. 1996, 241, 103–108. [Google Scholar] [CrossRef]
- Galimi, F.; Torti, D.; Sassi, F.; Isella, C.; Cora, D.; Gastaldi, S.; Ribero, D.; Muratore, A.; Massucco, P.; Siatis, D.; et al. Genetic and expression analysis of MET, MACC1, and HGF in metastatic colorectal cancer: Response to met inhibition in patient xenografts and pathologic correlations. Clin. Cancer Res. 2011, 17, 3146–3156. [Google Scholar] [CrossRef] [Green Version]
- Yushkevich, P.A.; Piven, J.; Hazlett, H.C.; Smith, R.G.; Ho, S.; Gee, J.C.; Gerig, G. User-guided 3D active contour segmentation of anatomical structures: Significantly improved efficiency and reliability. Neuroimage 2006, 31, 1116–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Griethuysen, J.J.M.; Fedorov, A.; Parmar, C.; Hosny, A.; Aucoin, N.; Narayan, V.; Beets-Tan, R.G.H.; Fillion-Robin, J.C.; Pieper, S.; Aerts, H. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Res. 2017, 77, e104–e107. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 26. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Farrell, A.C.; Jarzabek, M.A.; Lindner, A.U.; Carberry, S.; Conroy, E.; Miller, I.S.; Connor, K.; Shiels, L.; Zanella, E.R.; Lucantoni, F.; et al. Implementing Systems Modelling and Molecular Imaging to Predict the Efficacy of BCL-2 Inhibition in Colorectal Cancer Patient-Derived Xenograft Models. Cancers 2020, 12, 2978. https://doi.org/10.3390/cancers12102978
O’Farrell AC, Jarzabek MA, Lindner AU, Carberry S, Conroy E, Miller IS, Connor K, Shiels L, Zanella ER, Lucantoni F, et al. Implementing Systems Modelling and Molecular Imaging to Predict the Efficacy of BCL-2 Inhibition in Colorectal Cancer Patient-Derived Xenograft Models. Cancers. 2020; 12(10):2978. https://doi.org/10.3390/cancers12102978
Chicago/Turabian StyleO’Farrell, Alice C., Monika A. Jarzabek, Andreas U. Lindner, Steven Carberry, Emer Conroy, Ian S. Miller, Kate Connor, Liam Shiels, Eugenia R. Zanella, Federico Lucantoni, and et al. 2020. "Implementing Systems Modelling and Molecular Imaging to Predict the Efficacy of BCL-2 Inhibition in Colorectal Cancer Patient-Derived Xenograft Models" Cancers 12, no. 10: 2978. https://doi.org/10.3390/cancers12102978
APA StyleO’Farrell, A. C., Jarzabek, M. A., Lindner, A. U., Carberry, S., Conroy, E., Miller, I. S., Connor, K., Shiels, L., Zanella, E. R., Lucantoni, F., Lafferty, A., White, K., Meyer Villamandos, M., Dicker, P., Gallagher, W. M., Keek, S. A., Sanduleanu, S., Lambin, P., Woodruff, H. C., ... Prehn, J. H. M. (2020). Implementing Systems Modelling and Molecular Imaging to Predict the Efficacy of BCL-2 Inhibition in Colorectal Cancer Patient-Derived Xenograft Models. Cancers, 12(10), 2978. https://doi.org/10.3390/cancers12102978