KIR+ CD8+ T Lymphocytes in Cancer Immunosurveillance and Patient Survival: Gene Expression Profiling


 ,
,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
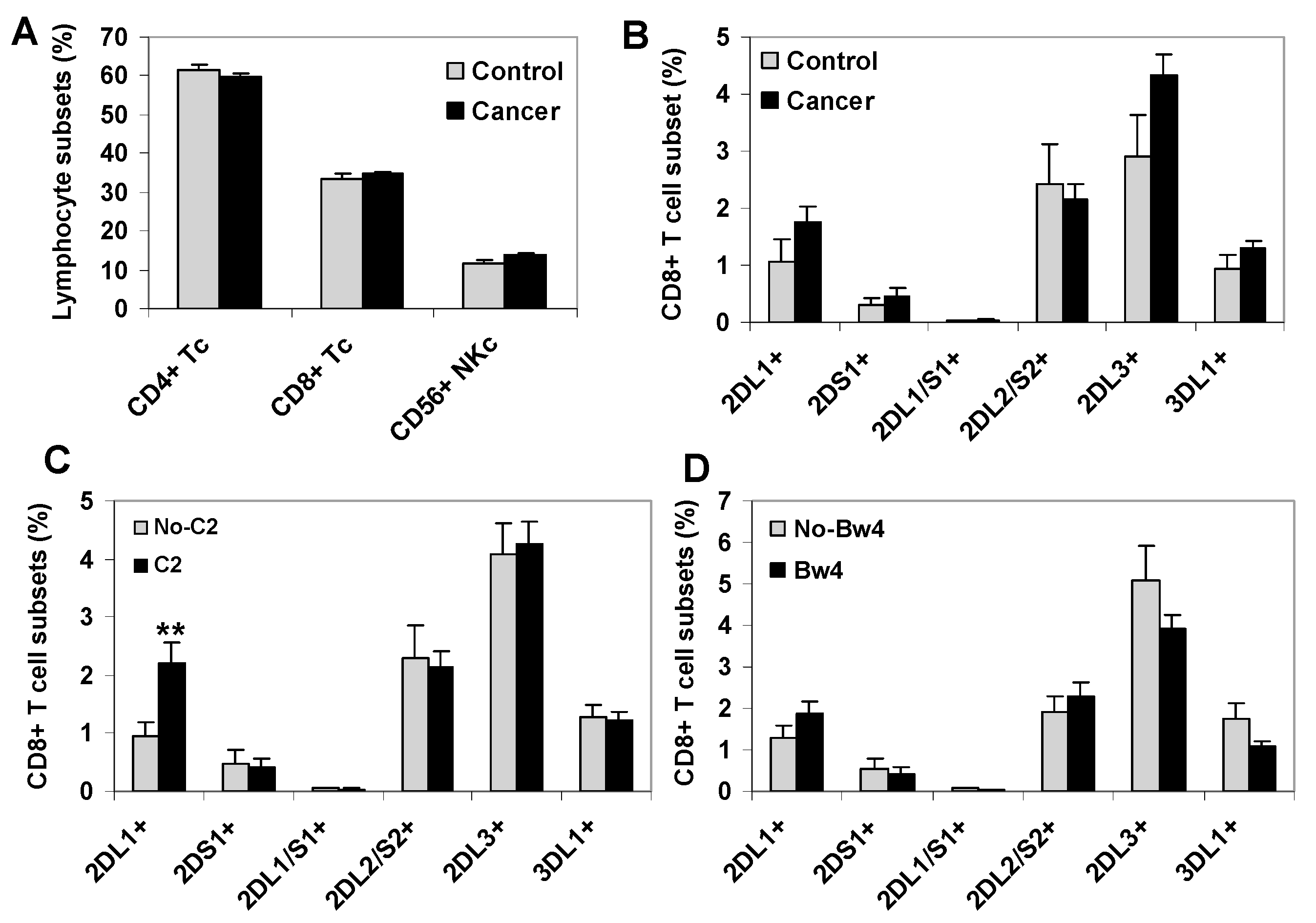
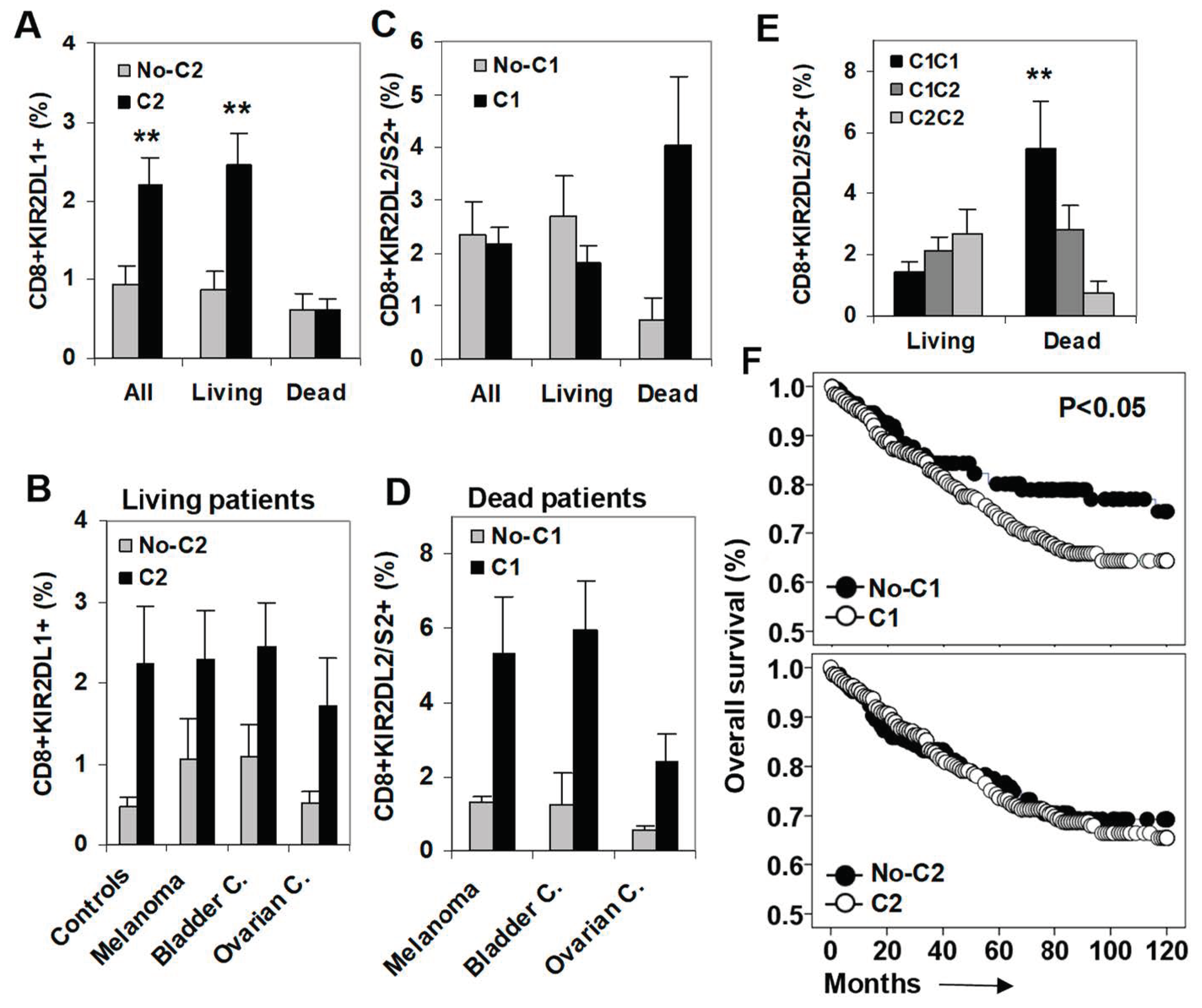
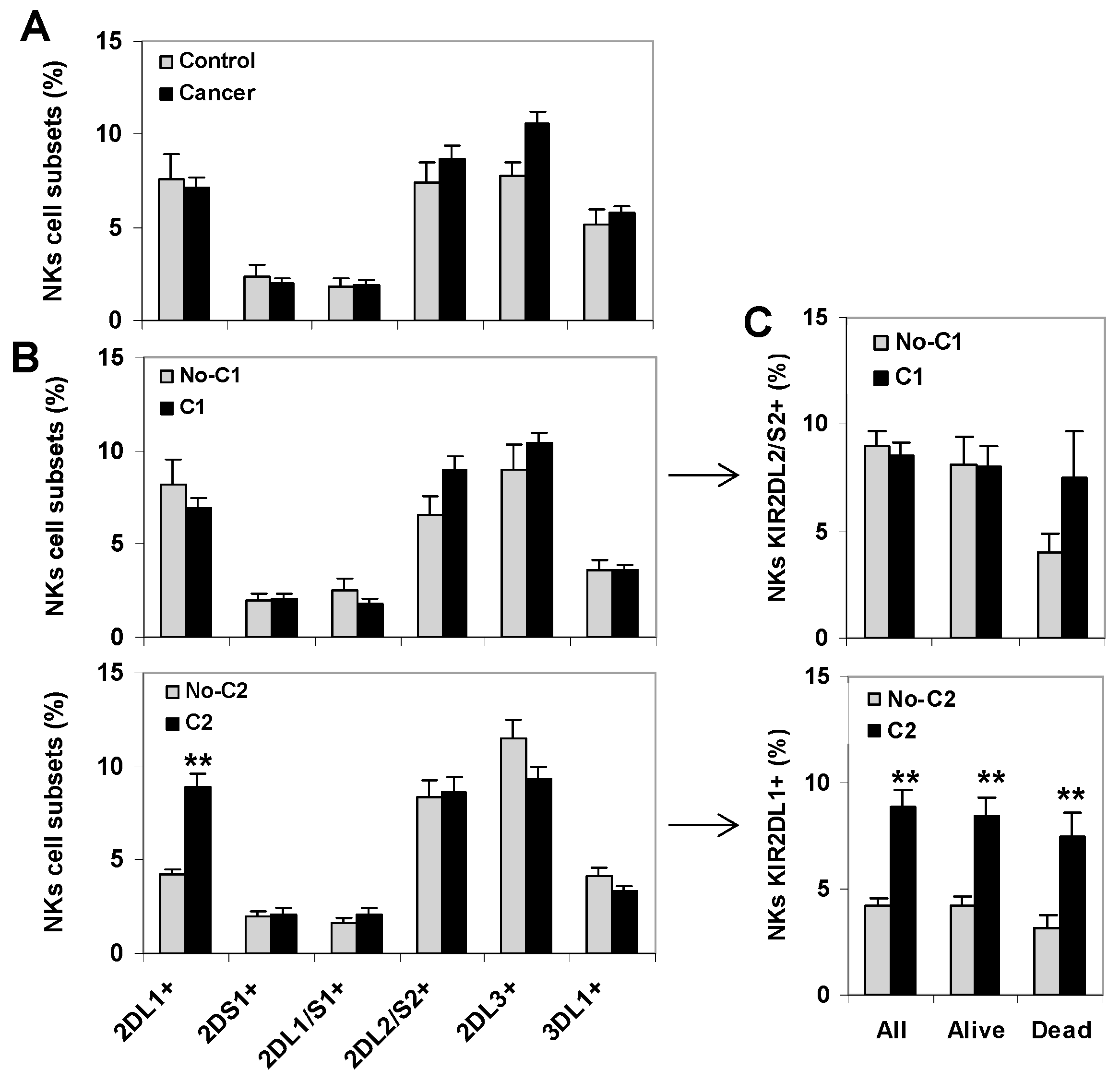
2.1. C2-Ligand Was Associated with Expansions of KIR2DL1+ CD8+ T Cells in Surviving Patients
2.2. C1-Ligand Was Associated with Dose-Dependent Expansion of KIR2DL2/S2+ CD8+ T Cells and Sorter Patient Survivals
2.3. KIR+ NK Cell Repertoires Were Not Associated to the Survival of Cancer Patients
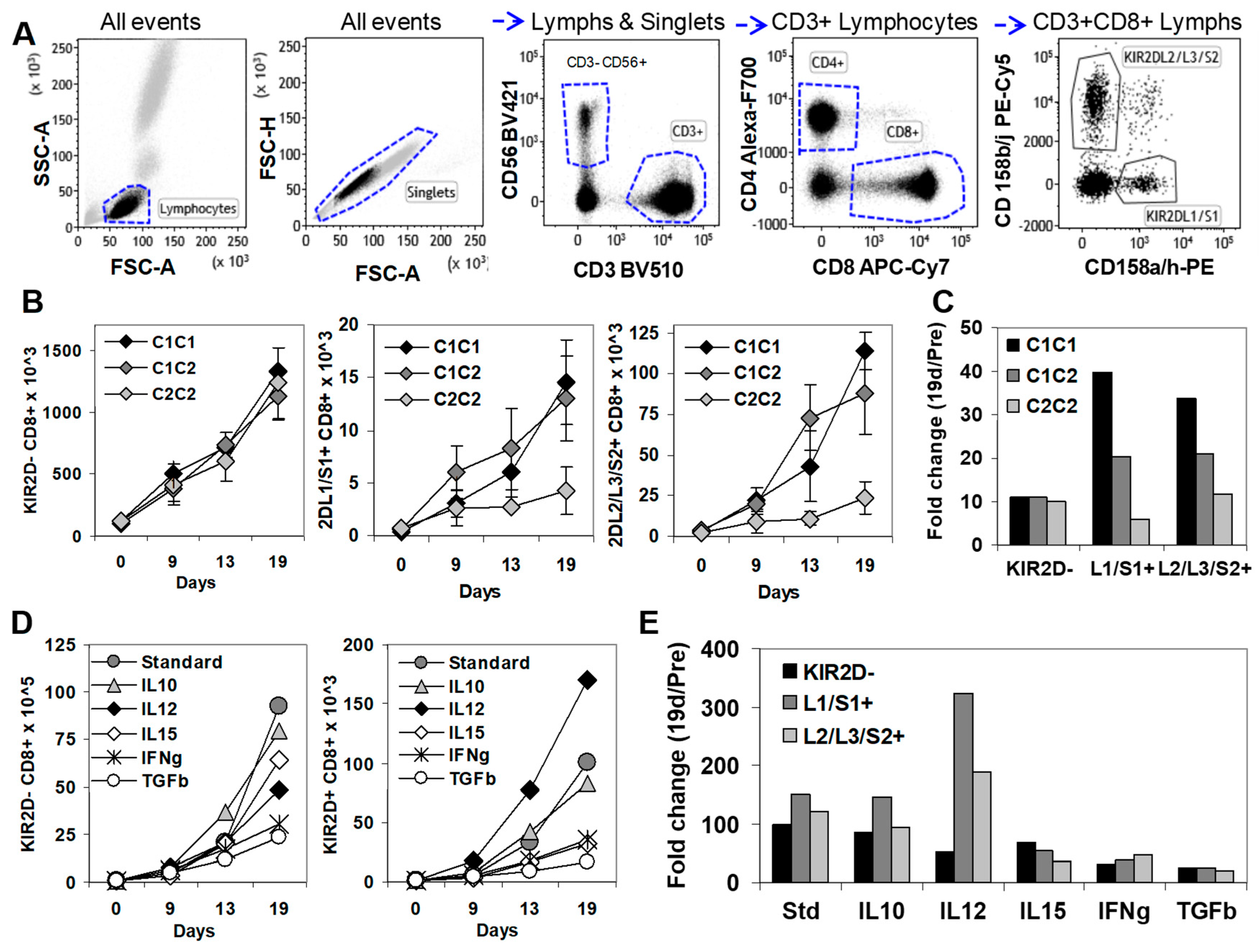
2.4. In Vitro, KIR2D+ CD8+ T Cells Expanded Preferentially in the Presence of HLA-C1 and IL-12
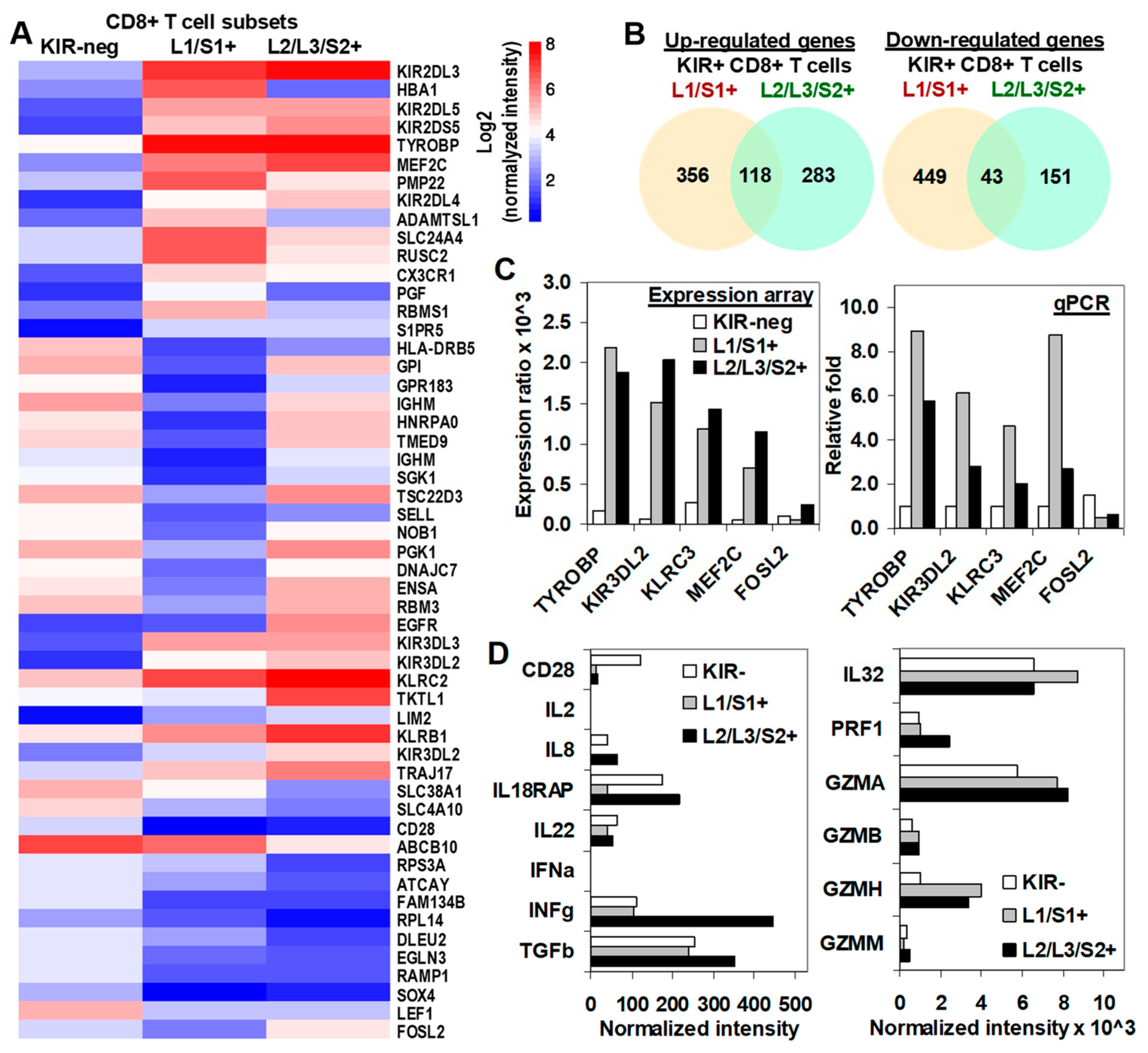
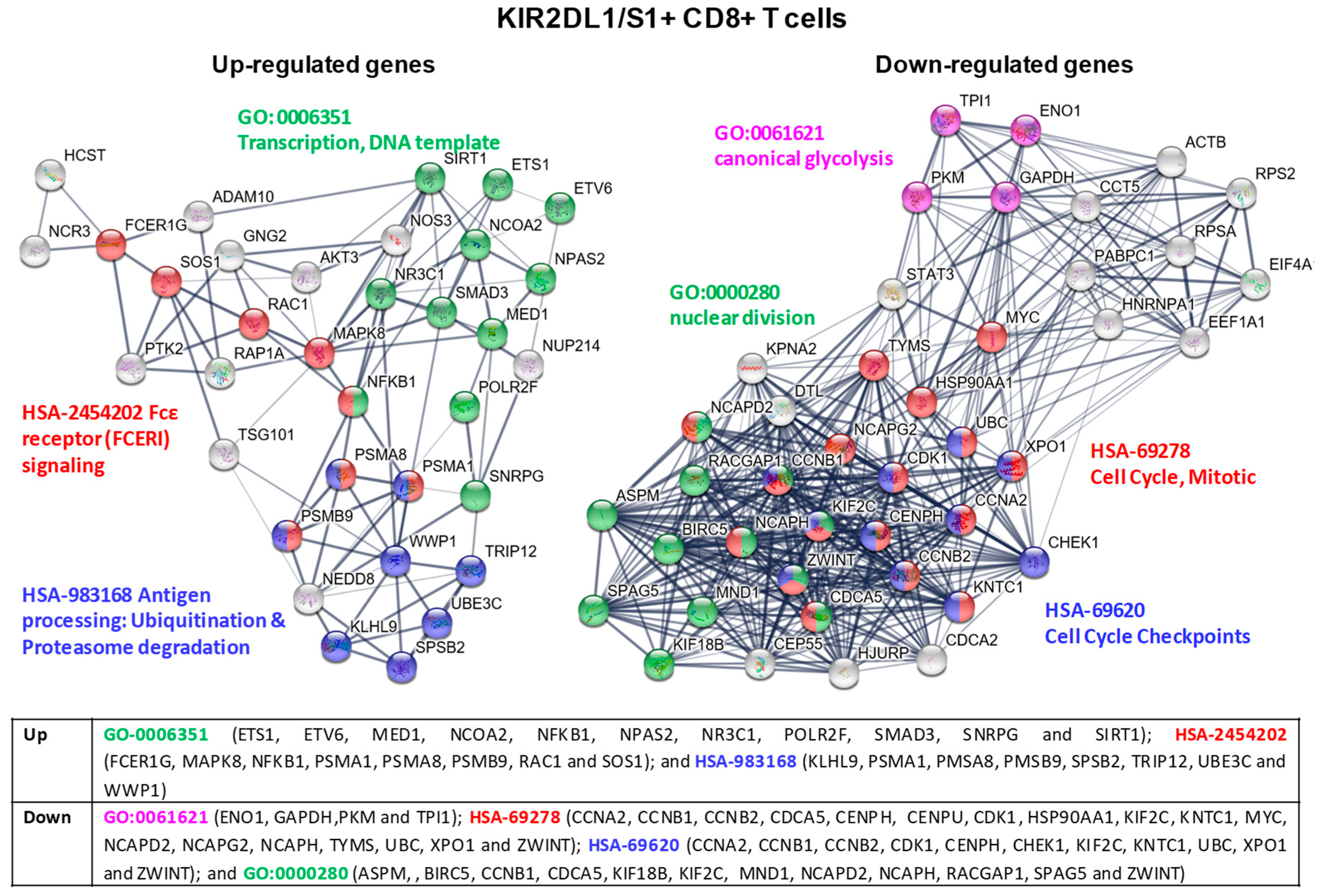
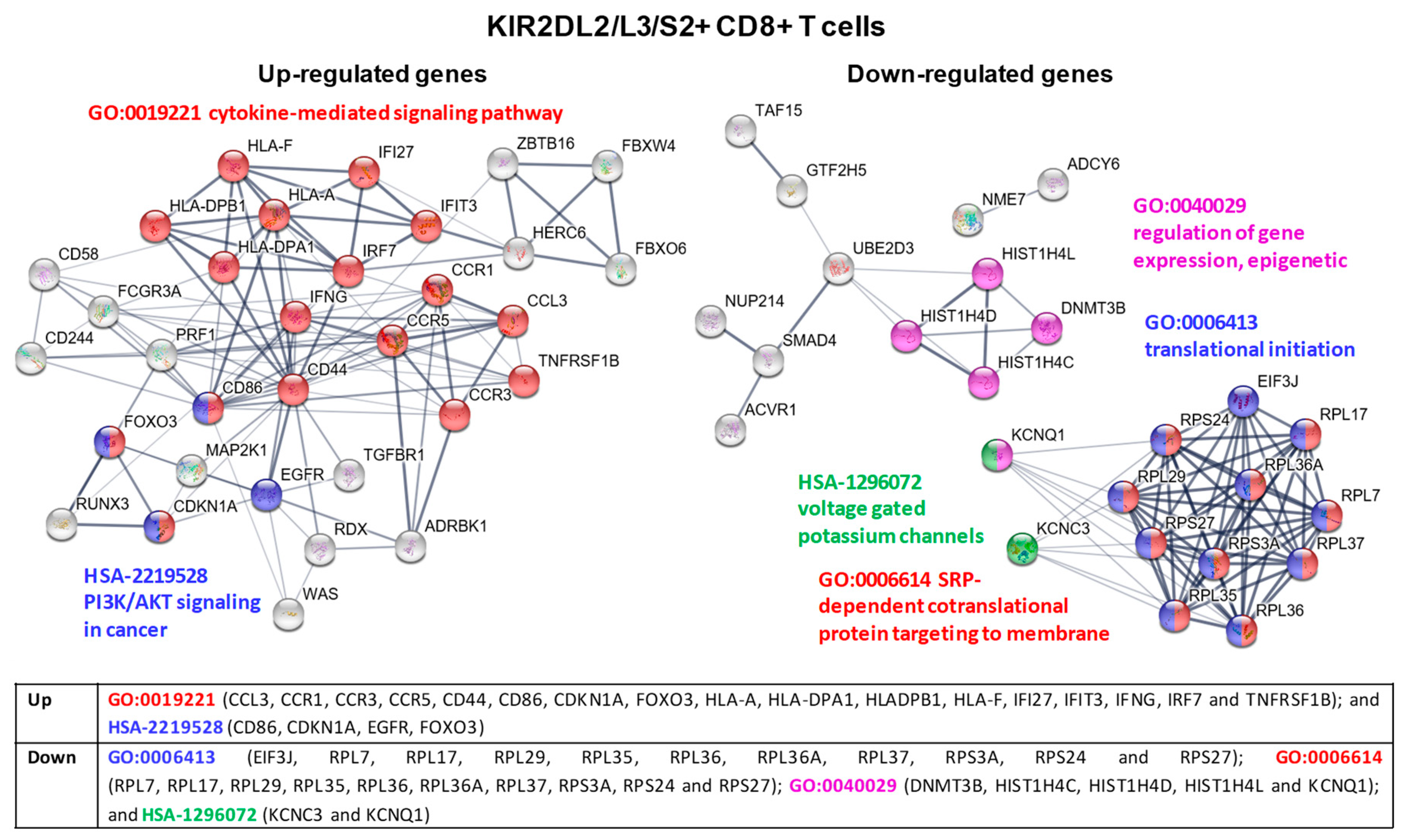
2.5. Transcriptional Profiling in KIR2D+ CD8+ T Cells: Functional Analysis
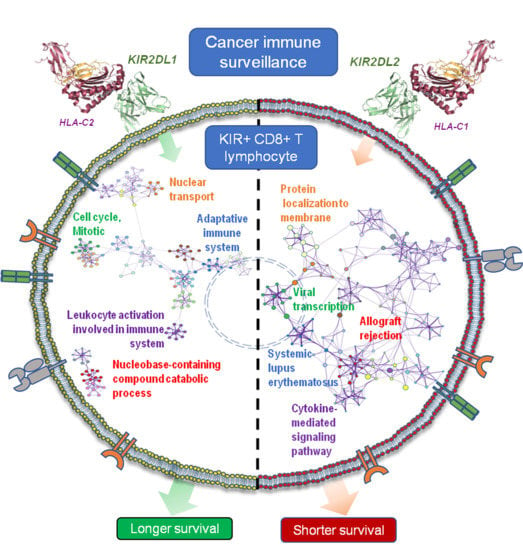
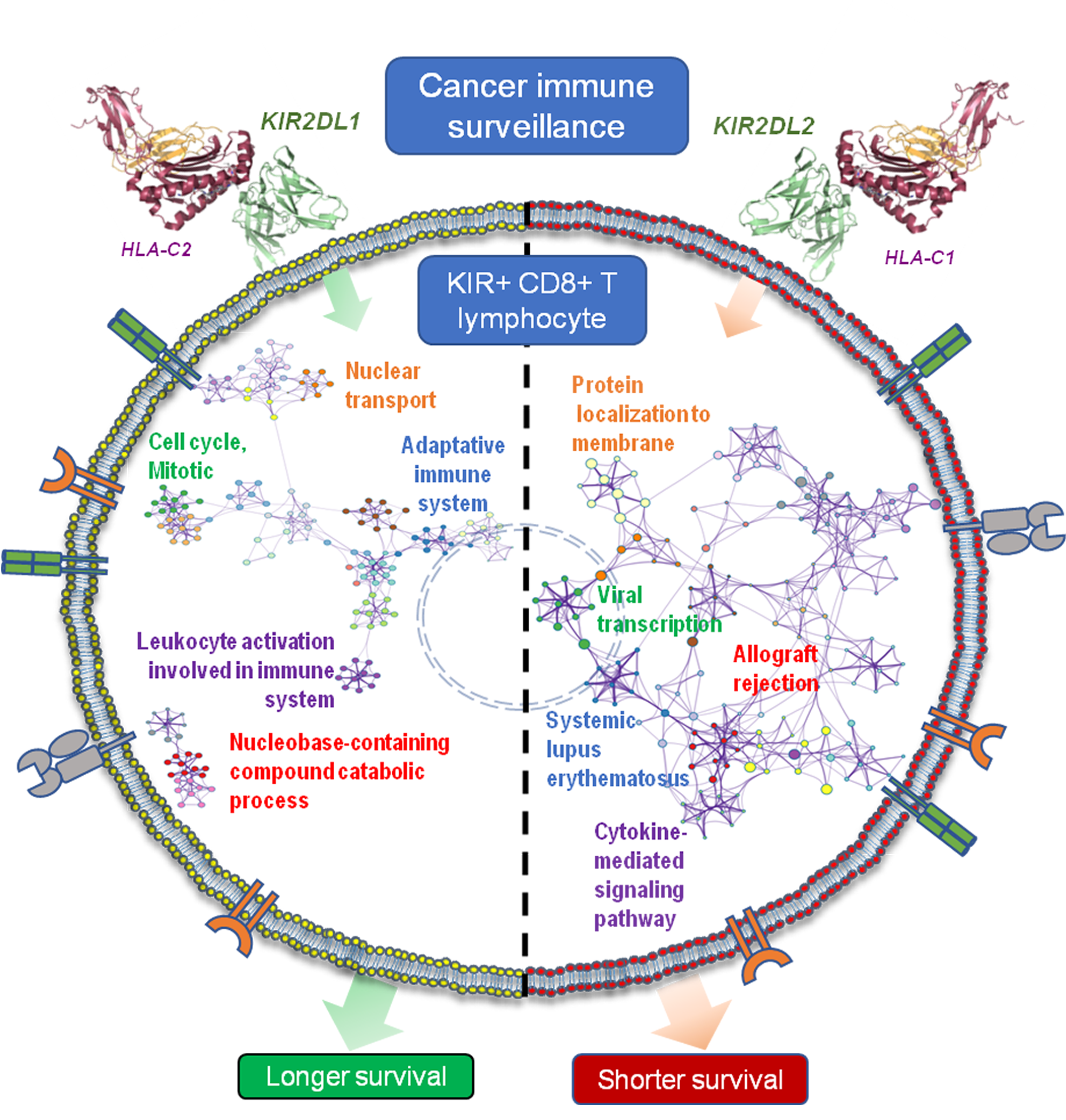
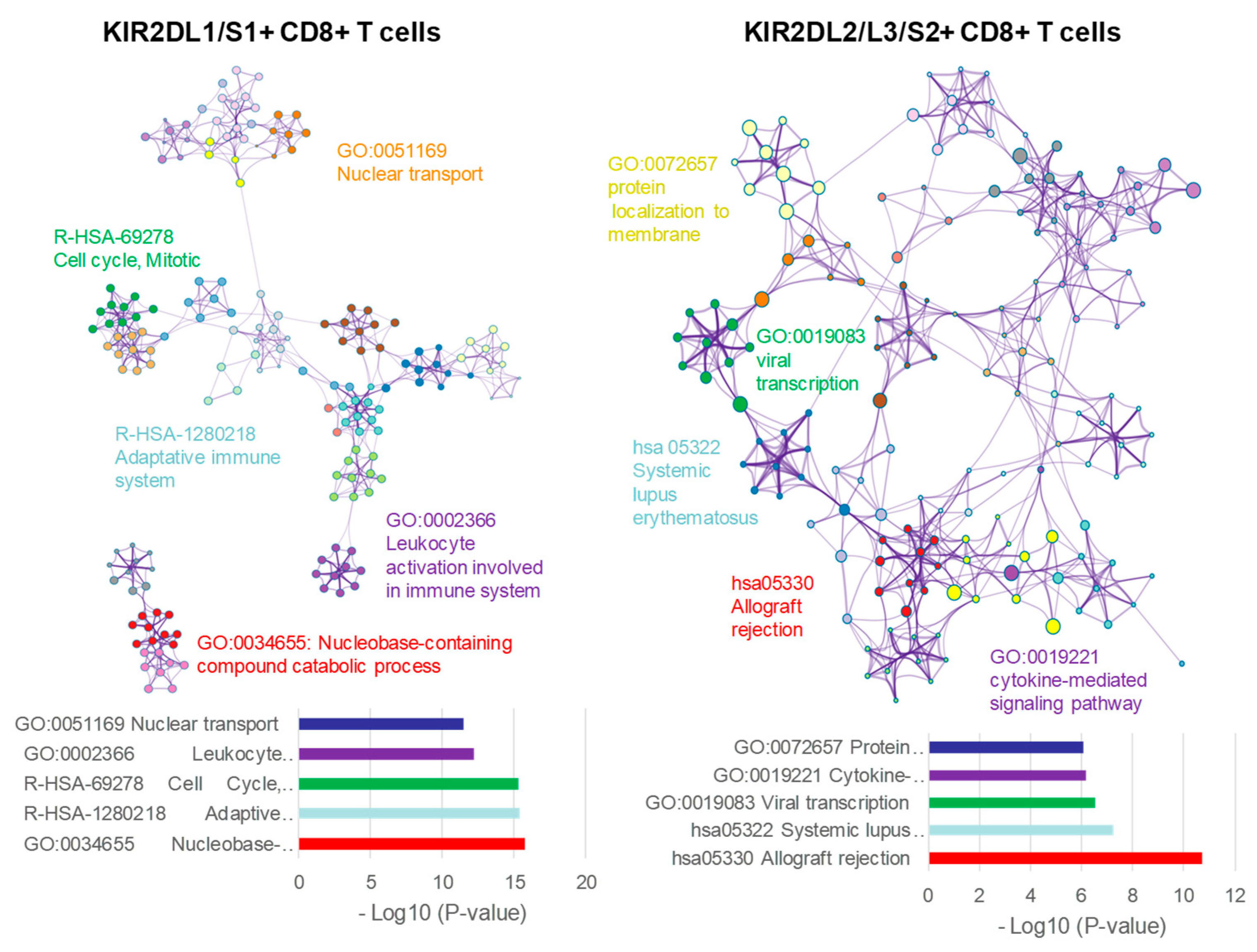
2.6. KIR2D+ CD8+ Functional Pathways
3. Discussion
4. Materials and Methods
4.1. Healthy Controls and Patient Characteristics
4.2. Immunophenotyping of CD8+ Peripheral Blood T Cells at Diagnosis
4.3. HLA-A, -B and -C and KIR Genotyping
4.4. In Vitro Expansion and Isolation of KIR2D−, KIR2DL1/S1+, and KIR2DL2/L3/S2+ CD8+ T Cells
4.5. RNA Extraction from KIR2D- and KIR2D+ CD8+ T Cells
4.6. T7 Amplification and Microarray Performance
4.7. Array Data Analysis
4.8. Real-Time Quantitative Reverse Transcription-PCR
4.9. Protein-Protein Interaction Network Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklós, I.; Toke, E.R.; Megyesi, M.; Molnar, L.; Tóth, J.; Lőrincz, O.; Somogyi, E.; Csiszovszki, Z.; Pántya, K.; Páles, P.; et al. Determination of the immunogenetic risk of developing cancer. J. Clin. Oncol. 2019, 37, e13132. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Born, C.; Bléry, M.; Steinle, A. MICAgen Mice Recapitulate the Highly Restricted but Activation-Inducible Expression of the Paradigmatic Human NKG2D Ligand MICA. Front. Immunol. 2020, 11, 960. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gao, F.; Li, N.; Li, Q.; Zhou, Y.; Yang, T.; Cai, Z.; Du, P.; Chen, F.; Cai, J. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am. J. Cancer Res. 2019, 9, 945–958. [Google Scholar]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Béziat, V.; Hilton, H.G.; Norman, P.J.; Traherne, J.A. Deciphering the killer-cell immunoglobulin-like receptor system at super-resolution for natural killer and T-cell biology. Immunology 2017, 150, 248–264. [Google Scholar] [CrossRef]
- Ohkawa, T.; Seki, S.; Dobashi, H.; Koike, Y.; Habu, Y.; Ami, K.; Hiraide, H.; Sekine, I. Systematic characterization of human CD8+ T cells with natural killer cell markers in comparison with natural killer cells and normal CD8+ T cells. Immunology 2001, 103, 281–290. [Google Scholar] [CrossRef]
- Arlettaz, L.; Degermann, S.; De Rham, C.; Roosnek, E.; Huard, B. Expression of inhibitory KIR is confined to CD8+ effector T cells and limits their proliferative capacity. Eur. J. Immunol. 2004, 34, 3413–3422. [Google Scholar] [CrossRef]
- Anfossi, N.; Doisne, J.-M.; Peyrat, M.-A.; Ugolini, S.; Bonnaud, O.; Bossy, D.; Pitard, V.; Merville, P.; Moreau, J.-F.; Delfraissy, J.-F.; et al. Coordinated Expression of Ig-Like Inhibitory MHC Class I Receptors and Acquisition of Cytotoxic Function in Human CD8 + T Cells. J. Immunol. 2004, 173, 7223–7229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, E.O. Regulation of immune responses through inhibitory receptors. Annu. Rev. Immunol. 1999, 17, 875–904. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, J.; Koning, F. The tortoise and the hare: Slowly evolving T-cell responses take hastily evolving KIR. Immunology 2010, 131, 301–309. [Google Scholar] [CrossRef]
- Campillo, J.A.; Martínez-Escribano, J.A.; Moya-Quiles, M.R.; Marín, L.A.; Muro, M.; Guerra, N.; Parrado, A.; Campos, M.; Frías, J.F.; Minguela, A.; et al. Natural killer receptors on CD8 T cells and natural killer cells from different HLA-C phenotypes in melanoma patients. Clin. Cancer Res. 2006, 12, 4822–4831. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Bhattacharjee, P.; Panda, A.K.; Kajal, K.; Bose, S.; Sa, G. Providence of the CD25+ KIR+ CD127- FOXP3- CD8+ T-cell subset determines the dynamics of tumor immune surveillance. Immunol. Cell Biol. 2018, 96, 1035–1048. [Google Scholar] [CrossRef]
- Boyton, R.J.; Altmann, D.M. Natural killer cells, killer immunoglobulin-like receptors and human leucocyte antigen class I in disease. Clin. Exp. Immunol. 2007, 149, 1–8. [Google Scholar] [CrossRef]
- Vierra-Green, C.; Roe, D.; Jayaraman, J.; Trowsdale, J.; Traherne, J.; Kuang, R.; Spellman, S.; Maiers, M. Estimating KIR haplotype frequencies on a cohort of 10,000 individuals: A comprehensive study on population variations, typing resolutions, and reference haplotypes. PLoS ONE 2016, 11, e0163973. [Google Scholar] [CrossRef] [Green Version]
- Guillamón, C.F.; Martínez-Sánchez, M.V.; Gimeno, L.; Campillo, J.A.; Server-Pastor, G.; Martínez-García, J.; Martínez-Escribano, J.; Torroba, A.; Ferri, B.; Abellán, D.J.; et al. Activating KIRs on Educated NK Cells Support Downregulation of CD226 and Inefficient Tumor Immunosurveillance. Cancer Immunol. Res. 2019, 7, 1307–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitahara, A.; Shimura, H.; Ito, A.; Tomiyama, K.; Ito, M.; Kawai, K. NKG2D ligation without T cell receptor engagement triggers both cytotoxicity and cytokine production in dendritic epidermal T cells. J. Investig. Dermatol. 2006, 126, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Guillamón, C.F.; Martínez-Sánchez, M.V.; Gimeno, L.; Mrowiec, A.; Martínez-García, J.; Server-Pastor, G.; Martínez-Escribano, J.; Torroba, A.; Ferri, B.; Abellán, D.; et al. NK Cell Education in Tumor Immune Surveillance: DNAM-1/KIR Receptor Ratios as Predictive Biomarkers for Solid Tumor Outcome. Cancer Immunol. Res. 2018, 6, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- Thiruchelvam-Kyle, L.; Hoelsbrekken, S.E.; Saether, P.C.; Bjørnsen, E.G.; Pende, D.; Fossum, S.; Daws, M.R.; Dissen, E. The Activating Human NK Cell Receptor KIR2DS2 Recognizes a β2-Microglobulin-Independent Ligand on Cancer Cells. J. Immunol. 2017, 198, 2556–2567. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.P.; Stojanovic, A.; Bauer, K.; Juraeva, D.; Tykocinski, L.-O.; Lorenz, H.-M.; Brors, B.; Cerwenka, A. Distinct human circulating NKp30+FcεRIγ+CD8+ T cell population exhibiting high natural killer-like antitumor potential. Proc. Natl. Acad. Sci. USA 2018, 115, E5980–E5989. [Google Scholar] [CrossRef] [Green Version]
- da Silva, R.F.; Petta, C.A.; Derchain, S.F.; Alici, E.; Guimarães, F. Up-regulation of DNAM-1 and NKp30, associated with improvement of NK cells activation after long-term culture of mononuclear cells from patients with ovarian neoplasia. Hum. Immunol. 2014, 75, 777–784. [Google Scholar] [CrossRef]
- Poltorak, M.; Meinert, I.; Stone, J.C.; Schraven, B.; Simeoni, L. Sos1 regulates sustained TCR-mediated Erk activation. Eur. J. Immunol. 2014, 44, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Llavero, F.; Artaso, A.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. Lck/PLCγ control migration and proliferation of interleukin (IL)-2-stimulated T cells via the Rac1 GTPase/glycogen phosphorylase pathway. Cell. Signal. 2016, 28, 1713–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, C.A.; Knudson, K.M.; Peng, B.J.; Teixeiro, E.; Daniels, M.A. The POSH/JIP-1 scaffold network regulates TCR-mediated JNK1 signals and effector function in CD8(+) T cells. Eur. J. Immunol. 2013, 43, 3361–3371. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Widjaja, C.E.; Olvera, J.G.; Metz, P.J.; Phan, A.T.; Savas, J.N.; de Bruin, G.; Leestemaker, Y.; Berkers, C.R.; de Jong, A.; Florea, B.I.; et al. Proteasome activity regulates CD8+ T lymphocyte metabolism and fate specification. J. Clin. Investig. 2017, 127, 3609–3623. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liao, L.; Park, Y.; Liu, Y.-C. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Ostuni, R. Adaptation and memory in immune responses. Nat. Immunol. 2019, 20, 783–792. [Google Scholar] [CrossRef]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Teh, S.-J.; Yang, J.; Chow, M.T.; Teh, H.-S. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J. Immunol. 2009, 183, 6051–6057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, K.; Ueda, Y.; Ozawa, M.; Matsuda, T.; Kinashi, T. Enhanced cytotoxic T-cell function and inhibition of tumor progression by Mst1 deficiency. FEBS Lett. 2016, 590, 68–75. [Google Scholar] [CrossRef]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment. Front. Immunol. 2018, 9, 2809. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Estess, P.; Siegelman, M. Bimolecular complex between rolling and firm adhesion receptors required for cell arrest; CD44 association with VLA-4 in T cell extravasation. Immunity 2004, 20, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.L.; Singh, N.P.; Nagarkatti, P.S.; Nagarkatti, M. CD44 mobilization in allogeneic dendritic cell-T cell immunological synapse plays a key role in T cell activation. J. Leukoc. Biol. 2008, 84, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Minguela, A.; Pastor, S.; Mi, W.; Richardson, J.A.; Ward, E.S. Feedback regulation of murine autoimmunity via dominant anti-inflammatory effects of interferon gamma. J. Immunol. 2007, 178, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Ohmatsu, H.; Humme, D.; Gonzalez, J.; Gulati, N.; Möbs, M.; Sterry, W.; Krueger, J.G. IL-32 induces indoleamine 2,3-dioxygenase+CD1c+ dendritic cells and indoleamine 2,3-dioxygenase+CD163+ macrophages: Relevance to mycosis fungoides progression. Oncoimmunology 2017, 6, e1181237. [Google Scholar] [CrossRef] [Green Version]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [Green Version]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: A KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Mandelboim, O.; Reyburn, H.T.; Valés-Gómez, M.; Pazmany, L.; Colonna, M.; Borsellino, G.; Strominger, J.L. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J. Exp. Med. 1996, 184, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Levings, M.K.; Sangregorio, R.; Sartirana, C.; Moschin, A.L.; Battaglia, M.; Orban, P.C.; Roncarolo, M.-G. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor beta, but not interleukin 10, and are distinct from type 1 T regulatory cells. J. Exp. Med. 2002, 196, 1335–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Patients (n = 249) | |||
|---|---|---|---|
| Melanoma (n = 80) | Bladder (n = 80) | Ovarian (n = 89) | |
| Sex (M/F, %M) 1 | 43/37 (53.7%) | 68/12 (85%) | 0/89 (0%) |
| Age (mean ± SD) | 60.6 ± 15.4 | 71.8 ± 10.2 | 58.7 ± 10.7 |
| Months since diagnosis (Mean ± SD) | 4.6 ± 14.5 | 14.9 ± 32.8 | 7.0 ± 19.6 |
| Histology (n) 2 | 35/45 | 36/44 | 69/20 |
| Staging (n) 3 | 51/29 | 2/78 | 26/63 |
| Treatment 4 | 10/9/61 | 51/20/9 | 32/49/8 |
| Progression (yes/no, %) | 27/80 (33.7%) | 24/80 (30.0%) | 37/89 (41.6%) |
| Death (yes/no, %) | 7/73 (8.7%) | 12/80 (15.0%) | 20/89 (22.5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimeno, L.; Serrano-López, E.M.; Campillo, J.A.; Cánovas-Zapata, M.A.; Acuña, O.S.; García-Cózar, F.; Martínez-Sánchez, M.V.; Martínez-Hernández, M.D.; Soto-Ramírez, M.F.; López-Cubillana, P.; et al. KIR+ CD8+ T Lymphocytes in Cancer Immunosurveillance and Patient Survival: Gene Expression Profiling. Cancers 2020, 12, 2991. https://doi.org/10.3390/cancers12102991
Gimeno L, Serrano-López EM, Campillo JA, Cánovas-Zapata MA, Acuña OS, García-Cózar F, Martínez-Sánchez MV, Martínez-Hernández MD, Soto-Ramírez MF, López-Cubillana P, et al. KIR+ CD8+ T Lymphocytes in Cancer Immunosurveillance and Patient Survival: Gene Expression Profiling. Cancers. 2020; 12(10):2991. https://doi.org/10.3390/cancers12102991
Chicago/Turabian StyleGimeno, Lourdes, Emilio M. Serrano-López, José A. Campillo, María A. Cánovas-Zapata, Omar S. Acuña, Francisco García-Cózar, María V. Martínez-Sánchez, María D. Martínez-Hernández, María F. Soto-Ramírez, Pedro López-Cubillana, and et al. 2020. "KIR+ CD8+ T Lymphocytes in Cancer Immunosurveillance and Patient Survival: Gene Expression Profiling" Cancers 12, no. 10: 2991. https://doi.org/10.3390/cancers12102991
APA StyleGimeno, L., Serrano-López, E. M., Campillo, J. A., Cánovas-Zapata, M. A., Acuña, O. S., García-Cózar, F., Martínez-Sánchez, M. V., Martínez-Hernández, M. D., Soto-Ramírez, M. F., López-Cubillana, P., Martínez-Escribano, J., Martínez-García, J., Corbalan-García, S., Álvarez-López, M. R., & Minguela, A. (2020). KIR+ CD8+ T Lymphocytes in Cancer Immunosurveillance and Patient Survival: Gene Expression Profiling. Cancers, 12(10), 2991. https://doi.org/10.3390/cancers12102991