Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies



Abstract
:Simple Summary
Abstract
1. Introduction
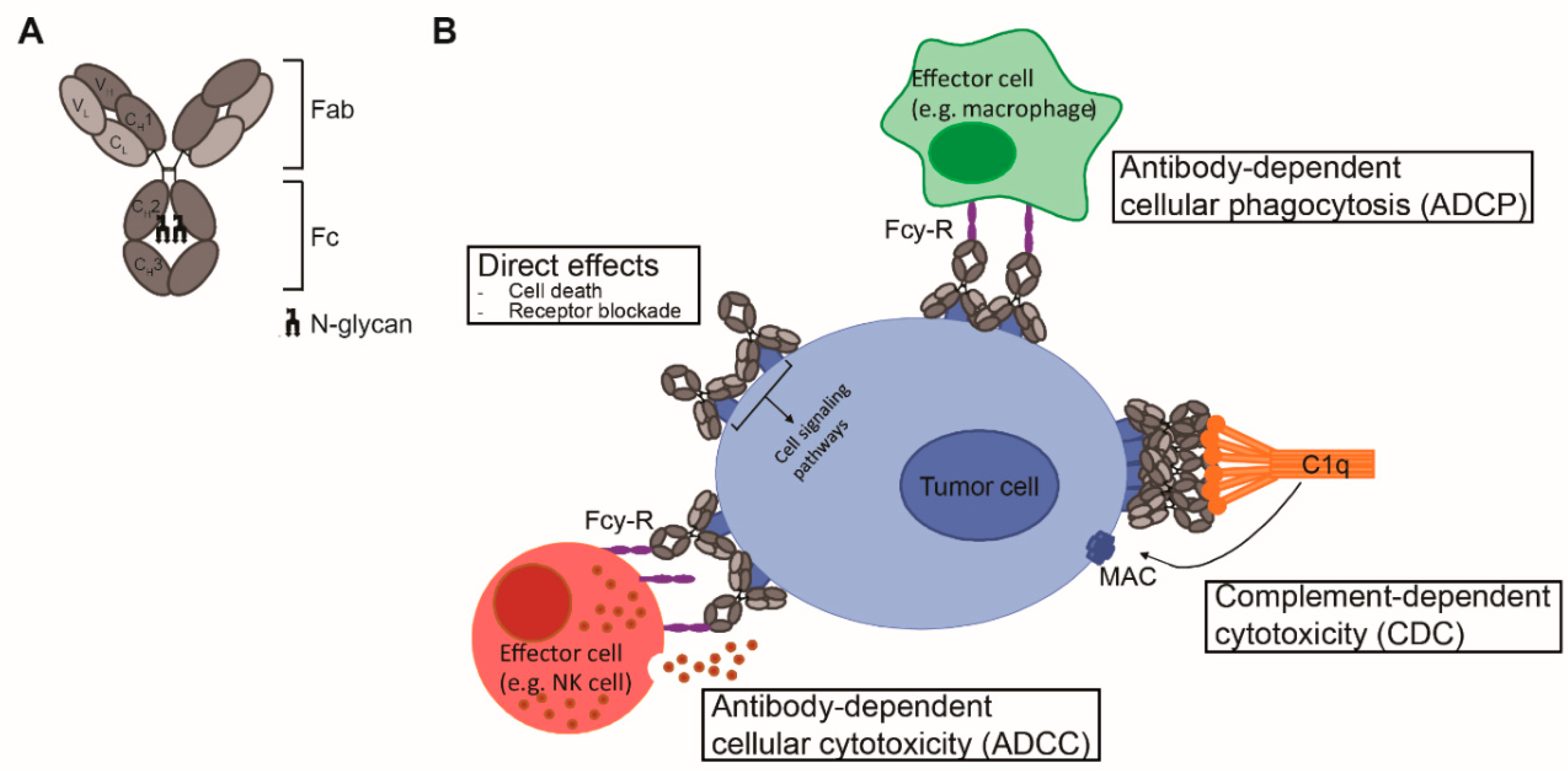
2. Antibody Structure
3. Fc-Effector Functions
3.1. ADCC/ADCP
3.2. CDC
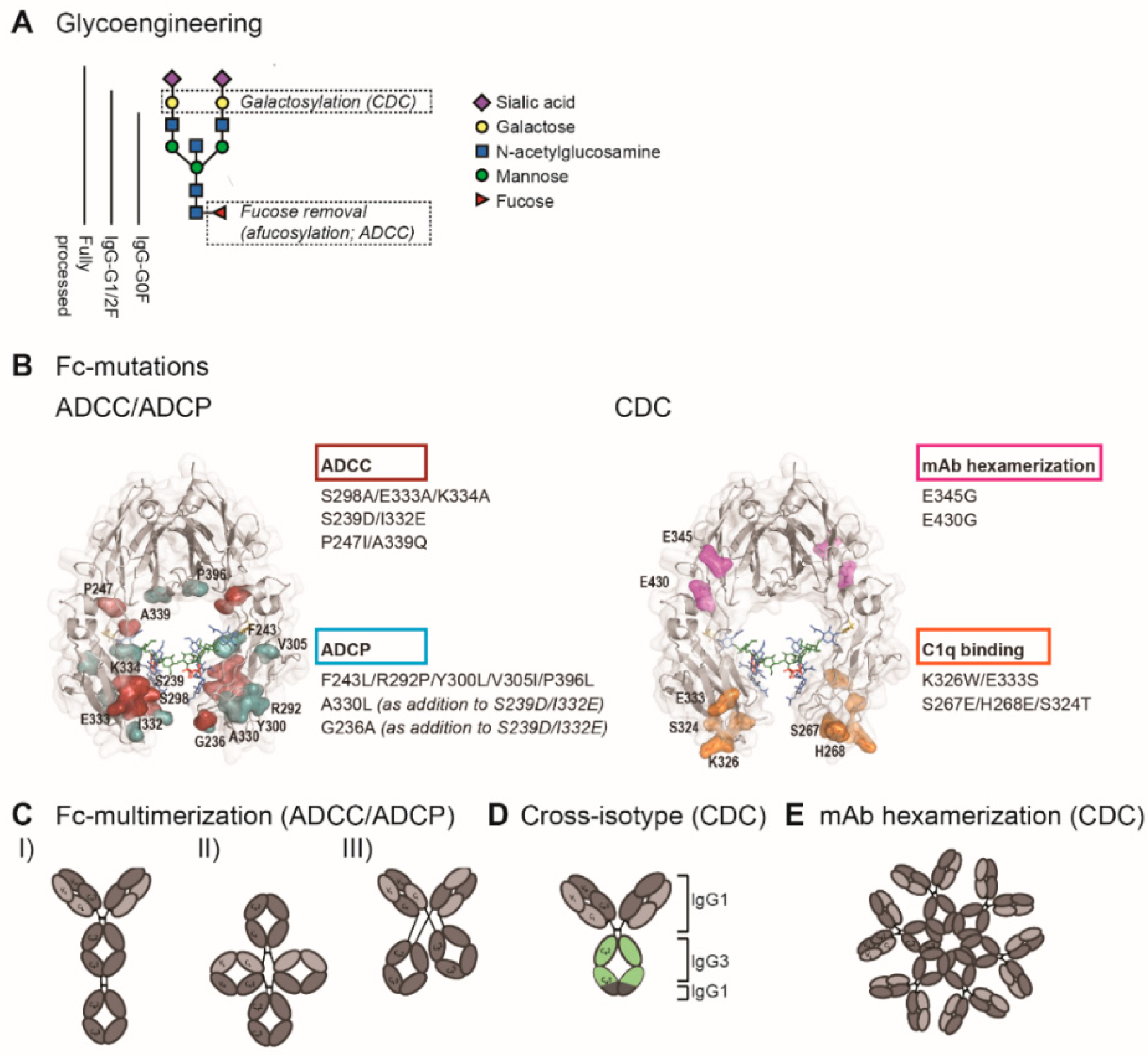
4. Fc Engineering to Enhance Fc-Effector Functions
4.1. Enhancing ADCC
4.1.1. Glycoengineering to Enhance FcγR Affinity
4.1.2. Site-Directed Mutagenesis to Enhance FcγR Affinity
4.1.3. Fc Multimerization
4.2. Enhancing ADCP
4.2.1. Glycoengineering to Enhance FcγR Affinity
4.2.2. Site-Directed Mutagenesis to Enhance FcγR Affinity
4.2.3. Fc Multimerization
4.3. Enhancing CDC
4.3.1. Glycoengineering to Enhance C1q Binding Affinity
4.3.2. Site-Directed Mutagenesis to Enhance C1q Binding Affinity
4.3.3. Antibody Hexamerization to Facilitate C1q Binding
4.3.4. Cross-Isotype Antibodies
5. Generation of Fc-Engineered mAbs
Glycoengineered mAbs
6. Clinical Experience with Fc-Engineered mAbs for B-Cell Malignancies
6.1. B-CLL and B-NHL
6.1.1. CD20
6.1.2. CD37
6.1.3. BAFF-R
6.1.4. CD19
6.2. Multiple Myeloma (MM)
6.2.1. CD38
6.2.2. HM1.24
6.2.3. ICAM-1
6.2.4. BCMA
7. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, T.J.; Osbourn, J.K.; Tempest, P.R. Human antibodies by design. Nat. Biotechnol. 1998, 16, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2019, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- De Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Brezski, R.J.; Georgiou, G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr. Opin. Immunol. 2016, 40, 62–69. [Google Scholar] [CrossRef]
- Swisher, J.F.A.; Feldman, G.M. The many faces of FcγRI: Implications for therapeutic antibody function. Immunol. Rev. 2015, 268, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Daëron, M. Fc receptor biology. Annu. Rev. Immunol. 1997, 15, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Ewang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, C.E.; Rose-Zerilli, M.J.J.; Machado, L.R.; Iriyama, C.; Hollox, E.J.; Cragg, M.S.; Strefford, J.C. Fcγ receptors: Genetic variation, function, and disease. Immunol. Rev. 2015, 268, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, P.A.; Van De Winkel, J.G.; Vlug, A.; Westerdaal, N.A.; Capel, P.J. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J. Immunol. 1991, 147, 1338–1343. [Google Scholar] [PubMed]
- Bruggeman, C.W.; Dekkers, G.; Bentlage, A.E.H.; Treffers, L.W.; Nagelkerke, S.Q.; Lissenberg-Thunnissen, S.; Koeleman, C.A.M.; Wuhrer, M.; Berg, T.K.V.D.; Rispens, T.; et al. Enhanced Effector Functions Due to Antibody Defucosylation Depend on the Effector Cell Fcγ Receptor Profile. J. Immunol. 2017, 199, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.-K.; Levy, R. Two Immunoglobulin G Fragment C Receptor Polymorphisms Independently Predict Response to Rituximab in Patients with Follicular Lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. Journal of clinical oncology. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2017, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Boyd, J.; Brampton, A.D.; Easterbrook-Smith, S.B.; Emanuel, E.J.; Novotny, J.; Rademacher, T.W.; Van Schravendijk, M.R.; Sternberg, M.J.E.; Dwek, R.A. The C1q receptor site on immunoglobulin G. Nature 1980, 288, 338–344. [Google Scholar] [CrossRef]
- Udaka, K.; Okada, M.; Utsumi, S. Co-operation between the pair of C gamma 2 domains in Clq-binding by rabbit IgG. Mol. Immunol. 1986, 23, 1103–1110. [Google Scholar] [CrossRef]
- Utsumi, S.; Okada, M.; Udaka, K.; Amano, T. Preparation and biologic characterization of fragments containing dimeric and monomeric C gamma 2 domain of rabbit IgG. Mol. Immunol. 1985, 22, 811–819. [Google Scholar] [CrossRef]
- Parce, J.W.; Kelley, D.; Heinzelmann, K. Measurement of antibody-dependent binding, proteolysis, and turnover of C1s on liposomal antigens localizes the fluidity-dependent step in C1 activation. Biochim. Biophys. Acta BBA Biomembr. 1983, 736, 92–98. [Google Scholar] [CrossRef]
- Hughes-Jones, N.C.; Gorick, B.D.; Howard, J.C.; Feinstein, A. Antibody density on rat red cells determines the rate of activation of the complement component Cl. Eur. J. Immunol. 1985, 15, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Teeling, J.L.; French, R.R.; Cragg, M.S.; Brakel, J.V.D.; Pluyter, M.; Huang, H.; Chan, C.; Parren, P.W.H.I.; Hack, C.E.; DeChant, M.; et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004, 104, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; De Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural Analysis of Human IgG-Fc Glycoforms Reveals a Correlation Between Glycosylation and Structural Integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Bowden, T.A.; Baruah, K.; Coles, C.H.; Harvey, D.J.; Yu, X.; Song, B.-D.; Stuart, D.I.; Aricescu, A.R.; Scanlan, C.N.; Jones, E.Y.; et al. Chemical and Structural Analysis of an Antibody Folding Intermediate Trapped during Glycan Biosynthesis. J. Am. Chem. Soc. 2012, 134, 17554–17563. [Google Scholar] [CrossRef]
- Jefferis, R. Isotype and glycoform selection for antibody therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-mediated effector functions: Molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef]
- Mimura, Y.; Sondermann, P.; Ghirlando, R.; Lund, J.; Young, S.P.; Goodall, M.; Jefferis, R. Role of oligosaccharide residues of IgG1-Fc in Fc gamma RIIb binding. J. Biol. Chem. 2001, 276, 45539–45547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, G.P.; Barb, A. The Structural Role of Antibody N-Glycosylation in Receptor Interactions. Structure 2015, 23, 1573–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M.R.; Lund, J.; Thompson, K.M.; Jefferis, R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing FcγRI and/or FcγRII receptors. Biochem. J. 1989, 259, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Kwon, H.S.; Lee, K.-H.; Lee, J.C.; Jung, S.T. Engineered aglycosylated full-length IgG Fc variants exhibiting improved FcγRIIIa binding and tumor cell clearance. mAbs 2017, 10, 278–289. [Google Scholar] [CrossRef]
- Yoon, H.W.; Jo, M.; Ko, S.; Kwon, H.S.; Lim, C.S.; Ko, B.J.; Lee, J.C.; Jung, S.T. Optimal combination of beneficial mutations for improved ADCC effector function of aglycosylated antibodies. Mol. Immunol. 2019, 114, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20167–20172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [Green Version]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or BisectingN-Acetylglucosamine of Human IgG1 Complex-type Oligosaccharides Shows the Critical Role of Enhancing Antibody-dependent Cellular Cytotoxicity. J. Biol. Chem. 2002, 278, 3466–3473. [Google Scholar] [CrossRef] [Green Version]
- Cambay, F.; Forest-Nault, C.; Dumoulin, L.; Seguin, A.; Henry, O.; Durocher, Y.; De Crescenzo, G. Glycosylation of Fcγ receptors influences their interaction with various IgG1 glycoforms. Mol. Immunol. 2020, 121, 144–158. [Google Scholar] [CrossRef]
- Falconer, D.J.; Subedi, G.P.; Marcella, A.M.; Barb, A. Antibody Fucosylation Lowers the FcγRIIIa/CD16a Affinity by Limiting the Conformations Sampled by the N162-Glycan. ACS Chem. Biol. 2018, 13, 2179–2189. [Google Scholar] [CrossRef]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brünker, P.; Umaña, P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakae, Y.; Satoh, T.; Yagi, H.; Yanaka, S.; Yamaguchi, T.; Isoda, Y.; Iida, S.; Okamoto, Y.; Kato, K. Conformational effects of N-glycan core fucosylation of immunoglobulin G Fc region on its interaction with Fcγ receptor IIIa. Sci. Rep. 2017, 7, 13780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomann, M.; Schlothauer, T.; Dashivets, T.; Malik, S.; Avenal, C.; Bulau, P.; Rüger, P.; Reusch, D. In Vitro Glycoengineering of IgG1 and Its Effect on Fc Receptor Binding and ADCC Activity. PLoS ONE 2015, 10, e0134949. [Google Scholar] [CrossRef]
- Thomann, M.; Reckermann, K.; Reusch, D.; Prasser, J.; Tejada, M.L. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol. Immunol. 2016, 73, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Peng, Y.; Berkowitz, S.A.; Engen, J.R. Post-translational Modifications Differentially Affect IgG1 Conformation and Receptor Binding. Mol. Cell. Proteom. 2010, 9, 1716–1728. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; De Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front. Immunol. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Crispin, M.; Pritchard, L.; Robinson, H.; Gorny, M.K.; Yu, X.; Bailey-Kellogg, C.; Ackerman, M.E.; Scanlan, C.; Zolla-Pazner, S.; et al. Identification of antibody glycosylation structures that predict monoclonal antibody Fc-effector function. AIDS 2014, 28, 2523–2530. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; DiLillo, D.J.; Bournazos, S.; Giddens, J.P.; Ravetch, J.V.; Wang, L.-X. Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. USA 2017, 114, 3485–3490. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Caaveiro, J.M.M.; Kiyoshi, M.; Tsumoto, K. Structural analysis of Fc/FcγR complexes: A blueprint for antibody design. Immunol. Rev. 2015, 268, 201–221. [Google Scholar] [CrossRef]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheney, C.M.; Stephens, D.M.; Mo, X.; Rafiq, S.; Butchar, J.; Flynn, J.M.; A Jones, J.; Maddocks, K.; O’Reilly, A.; Ramachandran, A.; et al. Ocaratuzumab, an Fc-engineered antibody demonstrates enhanced antibody-dependent cell-mediated cytotoxicity in chronic lymphocytic leukemia. mAbs 2014, 6, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimoto, F.; Igawa, T.; Kuramochi, T.; Katada, H.; Kadono, S.; Kamikawa, T.; Shida-Kawazoe, M.; Hattori, K. Novel asymmetrically engineered antibody Fc variant with superior FcgammaR binding affinity and specificity compared with afucosylated Fc variant. mAbs 2013, 5, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Gunasekaran, K.; Wang, W.; Razinkov, V.; Sekirov, L.; Leng, E.; Sweet, H.; Foltz, I.; Howard, M.; Rousseau, A.-M.; et al. Asymmetrical Fc Engineering Greatly Enhances Antibody-dependent Cellular Cytotoxicity (ADCC) Effector Function and Stability of the Modified Antibodies. J. Biol. Chem. 2014, 289, 3571–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, H.; Tezuka, T.; Tsuchida, W.; Maeda, H.; Kohroki, J.; Masuho, Y. Tandemly repeated Fc domain augments binding avidities of antibodies for Fcgamma receptors, resulting in enhanced antibody-dependent cellular cytotoxicity. Mol. Immunol. 2008, 45, 2752–2763. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Pelletier, M.; Cvitkovic, R.; Bonnell, J.; Chang, C.-Y.; Koksal, A.C.; O’Connor, E.; Gao, X.; Yu, X.-Q.; et al. Enhancement of antibody functions through Fc multiplications. mAbs 2017, 9, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulet, D.R.; Zwolak, A.; Williams, J.A.; Chiu, M.L.; Atkins, W.M. Design and characterization of novel dual Fc antibody with enhanced avidity for Fc receptors. Proteins Struct. Funct. Bioinform. 2020, 88, 689–697. [Google Scholar] [CrossRef]
- Sustmann, C.; Dickopf, S.; Regula, J.T.; Kettenberger, H.; Mølhøj, M.; Gassner, C.; Weininger, D.; Fenn, S.; Manigold, T.; Kling, L.; et al. DuoMab: A novel CrossMab-based IgG-derived antibody format for enhanced antibody-dependent cell-mediated cytotoxicity. mAbs 2019, 11, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Tada, M.; Aoyama, M.; Ishii-Watabe, A. Fcγ Receptor Activation by Human Monoclonal Antibody Aggregates. J. Pharm. Sci. 2020, 109, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Shibata-Koyama, M.; Iida, S.; Misaka, H.; Mori, K.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated rituximab potentiates human neutrophil phagocytosis through its high binding for FcgammaRIIIb and MHC class II expression on the phagocytotic neutrophils. Exp. Hematol. 2009, 37, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Birk, M.C.; Klein, C.; Gerdes, C.; Umana, P.; Bacac, M. Glycoengineering of Therapeutic Antibodies Enhances Monocyte/Macrophage-Mediated Phagocytosis and Cytotoxicity. J. Immunol. 2014, 192, 2252–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, C.T.; Veri, M.-C.; Gorlatov, S.; Tuaillon, N.; Burke, S.; Huang, L.; Inzunza, H.D.; Li, H.; Thomas, S.; Johnson, S.; et al. CD32B, the human inhibitory Fc-γ receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood 2006, 108, 2384–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Vihj, S.; Johnson, S.; Bonvini, E. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007, 67, 8882–8890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, J.O.; Karki, S.; Lazar, G.A.; Chen, H.; Dang, W.; Desjarlais, J.R. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 2008, 7, 2517–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Keremane, S.R.; Vielmetter, J.; Bjorkman, P.J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. J. Struct. Biol. 2016, 194, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, H.; Ootsubo, M.; Fukazawa, M.; Motoi, S.; Konakahara, S.; Masuho, Y. Enhanced antibody-dependent cellular phagocytosis by chimeric monoclonal antibodies with tandemly repeated Fc domains. J. Biosci. Bioeng. 2011, 111, 391–396. [Google Scholar] [CrossRef]
- Quast, I.; Keller, C.W.; Maurer, M.A.; Giddens, J.; Tackenberg, B.; Wang, L.-X.; Munz, C.; Nimmerjahn, F.; Dalakas, M.C.; Lünemann, J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Investig. 2015, 125, 4160–4170. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Matsui, M.; Kawasaki, N. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. mAbs 2018, 11, 350–372. [Google Scholar] [CrossRef]
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 2017, 8, 646. [Google Scholar] [CrossRef]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of Recombinant Monoclonal Antibody Effector Functions by Fc N-Glycan Remodeling in Vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; De Jong, R.N.; Bremer, E.T.V.D.; Beurskens, F.J.; Labrijn, A.F.; Ugurlar, D.; Gros, P.; Schuurman, J.; Parren, P.W.; Heck, A.J.R. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol. Cell 2016, 63, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idusogie, E.E.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Wong, P.Y.; Ultsch, M.; Meng, Y.G.; Mulkerrin, M.G. Mapping of the C1q Binding Site on Rituxan, a Chimeric Antibody with a Human IgG1 Fc. J. Immunol. 2000, 164, 4178–4184. [Google Scholar] [CrossRef] [Green Version]
- Thommesen, J.E.; Michaelsen, T.E.; Løset, G.Å.; Sandlie, I.; Brekke, O.H. Lysine 322 in the human IgG3 CH2 domain is crucial for antibody dependent complement activation. Mol. Immunol. 2000, 37, 995–1004. [Google Scholar] [CrossRef]
- Ugurlar, D.; Howes, S.C.; De Kreuk, B.-J.; Koning, R.I.; De Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered Antibodies with Increased Activity to Recruit Complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Cook, K.E.; Damschroder, M.M.; Woods, R.M.; Wu, H. Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J. Immunol. 2006, 177, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- De Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; Van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, H.-G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef] [Green Version]
- Brüggemann, M.; Williams, G.T.; Bindon, C.I.; Clark, M.R.; Walker, M.R.; Jefferis, R.; Waldmann, H.; Neuberger, M.S. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 1987, 166, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; In, M.; Takamura, H.; Nakagawa, T.; Shimizu, Y.; Kitajima, K.; Wakitani, M.; Ohta, S.; Satoh, M.; Shitara, K.; et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008, 68, 3863–3872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, T.P.; Parekh, R.B.; Moellering, B.J.; Prior, C.P. Different culture methods lead to differences in glycosylation of a murine IgG monoclonal antibody. Biochem. J. 1992, 285, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müthing, J.; Kemminer, S.E.; Conradt, H.S.; Šagi, D.; Nimtz, M.; Kärst, U.; Peter-Katalinić, J. Effects of buffering conditions and culture pH on production rates and glycosylation of clinical phase I anti-melanoma mouse IgG3 monoclonal antibody R24. Biotechnol. Bioeng. 2003, 83, 321–334. [Google Scholar] [CrossRef]
- Yamane-Ohnuki, N.; Kinoshita, S.; Inoue-Urakubo, M.; Kusunoki, M.; Iida, S.; Nakano, R.; Wakitani, M.; Niwa, R.; Sakurada, M.; Uchida, K.; et al. Establishment ofFUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [Google Scholar] [CrossRef]
- Davies, J.; Jiang, L.; Pan, L.-Z.; LaBarre, M.J.; Anderson, D.; Reff, M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FCγRIII. Biotechnol. Bioeng. 2001, 74, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Umaña, P.; Jean–Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Yogo, R.; Yamaguchi, Y.; Watanabe, H.; Yagi, H.; Satoh, T.; Nakanishi, M.; Onitsuka, M.; Omasa, T.; Shimada, M.; Maruno, T.; et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B. Antigen binding allosterically promotes Fc receptor recognition. mAbs 2018, 11, 58–74. [Google Scholar] [CrossRef]
- Cragg, M.S.; Walshe, C.A.; Ivanov, A.O.; Glennie, M.J. The Biology of CD20 and Its Potential as a Target for mAb Therapy. Curr. Dir. Autoimmun. 2004, 8, 140–174. [Google Scholar] [CrossRef]
- Boross, P.; Leusen, J.H.W. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar] [PubMed]
- Cragg, M.S.; Morgan, S.M.; Chan, H.T.C.; Morgan, B.P.; Filatov, A.V.; Johnson, P.W.M.; French, R.R.; Glennie, M.J. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 2003, 101, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; Van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.E.; Davies, F.A.J.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.J.; Phillips, E.H.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab-Based Induction and Maintenance Prolongs Progression-Free Survival (PFS) in Patients with Previously Untreated Follicular Lymphoma: Primary Results of the Randomized Phase 3 GALLIUM Study. Blood 2016, 128, 6. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; De La Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. New Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Martelli, M.; Trněný, M.; Liu, W.; Bolen, C.R.; Knapp, A.; Sahin, D.; Sellam, G.; Vitolo, U. A randomized, open-label, Phase III study of obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-Cell lymphoma: Final analysis of GOYA. J. Hematol. Oncol. 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, U.; Trněný, M.; Belada, D.; Burke, J.M.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.; Hong, X.; et al. Obinutuzumab or Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Previously Untreated Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017, 35, 3529–3537. [Google Scholar] [CrossRef]
- De Romeuf, C.; Dutertre, C.A.; Le Garff-Tavernier, M.; Fournier, N.; Gaucher, C.; Glacet, A.; Jorieux, S.; Bihoreau, N.; Behrens, C.K.; Béliard, R.; et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br. J. Haematol. 2008, 140, 635–643. [Google Scholar] [CrossRef]
- Le Garff-Tavernier, M.; Herbi, L.; De Romeuf, C.; Nguyen-Khac, F.; Davi, F.; Grelier, A.; Boudjoghra, M.; Maloum, K.; Choquet, S.; Urbain, R.; et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia 2014, 28, 230–233. [Google Scholar] [CrossRef]
- Barth, M.J.; Pendurti, G.; Tsai, P.-C.; Mavis, C.; Klener, P.; Czuczman, M.S.; Hernandez-Ilizaliturri, F. Ofatumumab (OFA) Is a Novel Anti-CD20 Monoclonal Antibody (mAb) with Improved Anti-Tumor Activity in Vitro and in Vivo in Mantle Cell Lymphoma (MCL) Pre-Clinical Models. Blood 2012, 120, 2757. [Google Scholar] [CrossRef]
- Sawas, A.; Farber, C.M.; Schreeder, M.T.; Khalil, M.Y.; Mahadevan, D.; Deng, C.; Amengual, J.E.; Nikolinakos, P.G.; Kolesar, J.M.; Kuhn, J.G.; et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br. J. Haematol. 2017, 177, 243–253. [Google Scholar] [CrossRef]
- Sharman, J.P.; Farber, C.M.; Mahadevan, D.; Schreeder, M.T.; Brooks, H.D.; Kolibaba, K.S.; Fanning, S.; Klein, L.; Greenwald, D.R.; Sportelli, P.; et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: Results of a phase 2 trial. Br. J. Haematol. 2016, 176, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Brander, D.M.; Mato, A.R.; Ghosh, N.; Schuster, S.J.; Kambhampati, S.; Burke, J.M.; Lansigan, F.; Schreeder, M.T.; Lunin, S.D.; et al. Effect of adding ublituximab to ibrutinib on PFS, ORR, and MRD negativity in previously treated high-risk chronic lymphocytic leukemia: Final results of the GENUINE phase III study. J. Clin. Oncol. 2020, 38, 8022. [Google Scholar] [CrossRef]
- Persky, D.O.; Dornan, D.; Goldman, B.H.; Braziel, R.M.; Fisher, R.I.; Leblanc, M.; Maloney, D.G.; Press, O.W.; Miller, T.P.; Rimsza, L.M. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemotherapy but not chemotherapy alone. Haematologica 2012, 97, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Jung, H.D.; Kim, J.G.; Lee, J.-J.; Yang, D.-H.; Park, Y.H.; Do, Y.R.; Shin, H.-J.; Kim, M.K.; Hyun, M.S.; et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood 2006, 108, 2720–2725. [Google Scholar] [CrossRef] [Green Version]
- Bowles, J.A.; Wang, S.-Y.; Link, B.K.; Allan, B.; Beuerlein, G.; Campbell, M.-A.; Marquis, D.; Ondek, B.; Wooldridge, J.E.; Smith, B.J.; et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood 2006, 108, 2648–2654. [Google Scholar] [CrossRef] [Green Version]
- Forero-Torres, A.; De Vos, S.; Pohlman, B.L.; Pashkevich, M.; Cronier, D.M.; Dang, N.H.; Carpenter, S.P.; Allan, B.W.; Nelson, J.G.; Slapak, C.A.; et al. Results of a Phase 1 Study of AME-133v (LY2469298), an Fc-Engineered Humanized Monoclonal Anti-CD20 Antibody, in FcγRIIIa-Genotyped Patients with Previously Treated Follicular Lymphoma. Clin. Cancer Res. 2012, 18, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganjoo, K.N.; De Vos, S.; Pohlman, B.L.; Flinn, I.W.; Forero-Torres, A.; Enas, N.H.; Cronier, D.M.; Dang, N.H.; Foon, K.A.; Carpenter, S.P.; et al. Phase 1/2 Study of Ocaratuzumab, an Fc-Engineered Humanized Anti-CD20 Monoclonal Antibody, in Low-Affinity FcγRIIIa Patients with Previously Treated Follicular Lymphoma. Leuk. Lymphoma 2014, 56, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W.; Vose, J.M.; Kahl, B.S.; Brunvand, M.W.; Goy, A.; Kasamon, Y.L.; Burington, B.; Li, J.; Ho, W.; Cheson, B.D. A Phase I Study of PRO131921, a Novel Anti-CD20 Monoclonal Antibody in Patients with Relapsed/Refractory CD20+ Indolent NHL: Correlation Between Clinical Responses and AUC Pharmacokinetics. Blood 2009, 114, 3742. [Google Scholar] [CrossRef]
- Cartron, G.; Watier, H. Obinutuzumab: What is there to learn from clinical trials? Blood 2017, 130, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morschhauser, F.; Marlton, P.; Vitolo, U.; Lindén, O.; Seymour, J.F.; Crump, M.; Coiffier, B.; Foà, R.; Wassner, E.; Burger, H.-U.; et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann. Oncol. 2010, 21, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Rösner, T.; Derer, S.; Kellner, C.; DeChant, M.; Lohse, S.; Vidarsson, G.; Peipp, M.; Valerius, T. An IgG3 switch variant of rituximab mediates enhanced complement-dependent cytotoxicity against tumour cells with low CD20 expression levels. Br. J. Haematol. 2013, 161, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Schwanbeck, R.; Skof, A.; Rösner, T.; Jansen, M.; Räuchle, A.; Kretschmer, A.; Scheidig, A.; Leusen, J.H.W.; Derer, S.; Valerius, T. Abstract 4601: Mechanisms of action for therapeutic antibody variants of human IgG3 isotype: Enhancing the CDC activity of cetuximab and rituximab. Cancer Res. 2017, 77 (Suppl. S13), 4601. [Google Scholar] [CrossRef]
- Natsume, A.; Shimizu-Yokoyama, Y.; Satoh, M.; Shitara, K.; Niwa, R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 2009, 100, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Wirt, T.; Rosskopf, S.; Rösner, T.; Eichholz, K.M.; Kahrs, A.; Lutz, S.; Kretschmer, A.; Valerius, T.; Klausz, K.; Otte, A.; et al. An Fc Double-Engineered CD20 Antibody with Enhanced Ability to Trigger Complement-Dependent Cytotoxicity and Antibody-Dependent Cell-Mediated Cytotoxicity. Transfus. Med. Hemotherapy 2017, 44, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Schwartz-Albiez, R.; Dörken, B.; Hofmann, W.; Moldenhauer, G. The B cell-associated CD37 antigen (gp40-52). Structure and subcellular expression of an extensively glycosylated glycoprotein. J. Immunol. 1988, 140, 905–914. [Google Scholar]
- Deckert, J.; Park, P.U.; Chicklas, S.; Yi, Y.; Li, M.; Lai, K.C.; Mayo, M.F.; Carrigan, C.N.; Erickson, H.K.; Pinkas, J.; et al. A novel anti-CD37 antibody-drug conjugate with multiple anti-tumor mechanisms for the treatment of B-cell malignancies. Blood 2013, 122, 3500–3510. [Google Scholar] [CrossRef] [Green Version]
- Heider, K.-H.; Kiefer, K.; Zenz, T.; Volden, M.; Stilgenbauer, S.; Ostermann, E.; Baum, A.; Lamche, H.; Küpcü, Z.; Jacobi, A.; et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood 2011, 118, 4159–4168. [Google Scholar] [CrossRef] [Green Version]
- Betrian, S.; Ysebaert, L.; Heider, K.H.; Delord, J.P.; Fournié, J.J.; Quillet-Mary, A. Idelalisib improves CD37 antibody BI 836826 cytotoxicity against chemo-resistant /relapse-initiating CLL cells: A rationale for combination treatment. Blood Cancer J. 2016, 6, e496. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Schleinitz, T.A.; Eichhorst, B.; Lang, F.; Offner, F.; Rossi, J.-F.; Schroyens, W.; Neste, E.V.D.; Ysebaert, L.; Von Wangenheim, U.; et al. Phase 1 first-in-human trial of the anti-CD37 antibody BI 836826 in relapsed/refractory chronic lymphocytic leukemia. Leukemia 2019, 33, 2531–2535. [Google Scholar] [CrossRef] [PubMed]
- Kroschinsky, F.; Middeke, J.M.; Janz, M.; Lenz, G.; Witzens-Harig, M.; Bouabdallah, R.; La Rosée, P.; Viardot, A.; Salles, G.; Kim, S.J.; et al. Phase I dose escalation study of BI 836826 (CD37 antibody) in patients with relapsed or refractory B-cell non-Hodgkin lymphoma. Investig. New Drugs 2020. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; Van Der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; Brink, E.N.V.D.; Brakel, J.H.N.V.D.; Rademaker, H.J.; Van Kessel, B.; Noort, J.V.D.; et al. DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, H.J.; Oostindie, S.C. Potent preclinical efficacy of DuoHexaBody-CD37 in B-cell malignancies. Hemasphere 2020. under revision. [Google Scholar]
- Thompson, J.S.; Bixler, S.A.; Qian, F.; Vora, K.; Scott, M.L.; Cachero, T.G.; Hession, C.; Schneider, P.; Sizing, I.D.; Mullen, C.; et al. BAFF-R, a Newly Identified TNF Receptor That Specifically Interacts with BAFF. Science 2001, 293, 2108–2111. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, E.M.; Cheney, C.; Jones, J.A.; Flynn, J.M.M.; Maddocks, K.; Andritsos, L.A.; Huet, H.; Gram, H.; Baeck, J.; Muthusamy, N.; et al. B-1239, a Novel Anti-BAFF-R Afucosylated Human Antibody, Promotes Potent Natural Killer Cell- Mediated Antibody Dependent Cellular Cytotoxicity In Chronic Lymphocytic Leukemia Cells In- Vitro and Depletion Of Circulating Leukemic CLL B Cells In-Vivo. Blood 2013, 122, 4185. [Google Scholar] [CrossRef]
- McWilliams, E.M.; Lucas, C.R.; Chen, T.; Harrington, B.K.; Wasmuth, R.; Campbell, A.; Rogers, K.A.; Cheney, C.M.; Mo, X.; Andritsos, L.A.; et al. Anti–BAFF-R antibody VAY-736 demonstrates promising preclinical activity in CLL and enhances effectiveness of ibrutinib. Blood Adv. 2019, 3, 447–460. [Google Scholar] [CrossRef]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Danhof, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, R.H.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-Cell Depletion In Vitro and In Vivo with an Afucosylated Anti-CD19 Antibody. J. Pharmacol. Exp. Ther. 2010, 335, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Cardarelli, P.M.; Rao-Naik, C.; Chen, S.; Huang, H.; Pham, A.; Moldovan-Loomis, M.-C.; Pan, C.; Preston, B.; Passmore, D.; Liu, J.; et al. A nonfucosylated human antibody to CD19 with potent B-cell depletive activity for therapy of B-cell malignancies. Cancer Immunol. Immunother. 2010, 59, 257–265. [Google Scholar] [CrossRef]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent In vitro and In vivo Activity of an Fc-Engineered Anti-CD19 Monoclonal Antibody against Lymphoma and Leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmachi, K.; Ogura, M.; Suehiro, Y.; Ando, K.; Uchida, T.; Choi, I.; Ogawa, Y.; Kobayashi, M.; Fukino, K.; Yokoi, Y.; et al. A multicenter phase I study of inebilizumab, a humanized anti-CD19 monoclonal antibody, in Japanese patients with relapsed or refractory B-cell lymphoma and multiple myeloma. Int. J. Hematol. 2019, 109, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Awan, F.; Flinn, I.W.; Berdeja, J.G.; Wiley, E.; Mansoor, S.; Huang, Y.; Lozanski, G.; Foster, P.A.; Byrd, J.C. A phase 1 trial of the Fc-engineered CD19 antibody XmAb5574 (MOR00208) demonstrates safety and preliminary efficacy in relapsed CLL. Blood 2014, 124, 3553–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurczak, W.; Zinzani, P.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Salles, G.; Duell, J.; Barca, E.G.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Van De Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.; Janmaat, M.L.; Andringa, G.; Kil, L.; Van Kessel, B.; Frerichs, K.A.; Lingnau, A.; Freidig, A.; Mutis, T.; Sasser, A.K.; et al. Hexabody-CD38, a Novel CD38 Antibody with a Hexamerization Enhancing Mutation, Demonstrates Enhanced Complement-Dependent Cytotoxicity and Shows Potent Anti-Tumor Activity in Preclinical Models of Hematological Malignancies. Blood 2019, 134 (Suppl. S1), 3106. [Google Scholar] [CrossRef]
- Choudhury, A.; Ortiz, D.F.; Argueta, S.; Garofalo, K.; Lansing, J.C.; Jetley, U.; Wilkins, D.; Bosques, C.; Cochran, E.; Bhatnagar, N.; et al. Abstract 561: Discovery of a potential best-in-class anti-CD38 therapeutic utilizing Fc multimerization. Cancer Res. 2019, 79 (Suppl. S13). [Google Scholar] [CrossRef]
- Goto, T.; Kennel, S.J.; Abe, M.; Takishita, M.; Kosaka, M.; Solomon, A.; Saito, S. A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood 1994, 84, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Ohtomo, T.; Sugamata, Y.; Ozaki, Y.; Ono, K.; Yoshimura, Y.; Kawai, S.; Koishihara, Y.; Ozaki, S.; Kosaka, M.; Hirano, T.; et al. Molecular Cloning and Characterization of a Surface Antigen Preferentially Overexpressed on Multiple Myeloma Cells. Biochem. Biophys. Res. Commun. 1999, 258, 583–591. [Google Scholar] [CrossRef]
- Schliemann, C.; Roesli, C.; Kamada, H.; Borgia, B.; Fugmann, T.; Klapper, W.; Neri, D. In vivo biotinylation of the vasculature in B-cell lymphoma identifies BST-2 as a target for antibody-based therapy. Blood 2010, 115, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, S.; Osei, E.S.; Kaplan, D.; Chen, Y.H.; Meyerson, H.J. CD317 is over-expressed in B-cell chronic lymphocytic leukemia, but not B-cell acute lymphoblastic leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 1613–1621. [Google Scholar] [PubMed]
- Gramatzki, R.; Szabo, E.; Gramatzki, M.; Peipp, M.; Weller, M. Immu-54. Cd317 expression in human glioblastoma: A target for immunotherapy. Neuro Oncol. 2017, 19 (Suppl. S6), vi124–vi125. [Google Scholar] [CrossRef]
- Wang, W.; Nishioka, Y.; Ozaki, S.; Jalili, A.; Abe, S.; Kakiuchi, S.; Kishuku, M.; Minakuchi, K.; Matsumoto, T.; Sone, S. HM1.24 (CD317) is a novel target against lung cancer for immunotherapy using anti-HM1.24 antibody. Cancer Immunol. Immunother. 2009, 58, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-L.; Wu, L.; Yu, G.-T.; Zhang, W.-F.; Liu, B.; Sun, Z.-J. CD317 Signature in Head and Neck Cancer Indicates Poor Prognosis. J. Dent. Res. 2018, 97, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Kawai, S.; Habu, K.; Sugimoto, M.; Shiraiwa, H.; Iijima, S.; Ozaki, S.; Matsumoto, T.; Yamada-Okabe, H. A defucosylated anti-CD317 antibody exhibited enhanced antibody-dependent cellular cytotoxicity against primary myeloma cells in the presence of effectors from patients. Cancer Sci. 2010, 101, 2227–2233. [Google Scholar] [CrossRef]
- Harada, T.; Ozaki, S.; Oda, A.; Tsuji, D.; Ikegame, A.; Iwasa, M.; Udaka, K.; Fujii, S.; Nakamura, S.; Miki, H.; et al. Combination with a Defucosylated Anti-HM1.24 Monoclonal Antibody plus Lenalidomide Induces Marked ADCC against Myeloma Cells and Their Progenitors. PLoS ONE 2013, 8, e83905. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Horton, H.M.; Kong, S.-Y.; Pong, E.; Chen, H.; Cemerski, S.; Bernett, M.J.; Nguyen, D.-H.T.; Karki, S.; Chu, S.Y.; et al. Potent in vitro and in vivo activity of an Fc-engineered humanized anti-HM1.24 antibody against multiple myeloma via augmented effector function. Blood 2012, 119, 2074–2082. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Mörsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for Cell Adhesion-Mediated Drug Resistance of Multiple Myeloma Cells in Vivo. Int. J. Biol. Markers 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Sampaio, M.S.S.; Vettore, A.L.; Yamamoto, M.; Chauffaille, M.L.; Zago, M.A.; Colleoni, G.W.B. Expression of eight genes of nuclear factor-kappa B pathway in multiple myeloma using bone marrow aspirates obtained at diagnosis. Histol. Histopathol. 2009, 24, 991–997. [Google Scholar]
- Veitonmäki, N.; Hansson, M.; Zhan, F.; Sundberg, A.; Löfstedt, T.; Ljungars, A.; Li, Z.-C.; Martinsson-Niskanen, T.; Zeng, M.; Yang, Y.; et al. A Human ICAM-1 Antibody Isolated by a Function-First Approach Has Potent Macrophage-Dependent Antimyeloma Activity In Vivo. Cancer Cell 2013, 23, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, M.; Gimsing, P.; Badros, A.; Nahi, H.; Offner, F.; Salomo, M.; Sonesson, E.; Mau-Sørensen, M.; Stenberg, Y.; Sundberg, A.; et al. A Phase I Dose-Escalation Study of Antibody BI-505 in Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2015, 21, 2730–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichert, S.; Juliusson, G.; Johansson, Å.; Sonesson, E.; Teige, I.; Wickenberg, A.T.; Frendeus, B.; Korsgren, M.; Hansson, M. A single-arm, open-label, phase 2 clinical trial evaluating disease response following treatment with BI-505, a human anti-intercellular adhesion molecule-1 monoclonal antibody, in patients with smoldering multiple myeloma. PLoS ONE 2017, 12, e0171205. [Google Scholar] [CrossRef] [PubMed]
- Klausz, K.; Cieker, M.; Kellner, C.; Oberg, H.H.; Kabelitz, D.; Valerius, T.; Burger, R.; Gramatzki, M.; Peipp, M. A novel Fc-engineered human ICAM-1/CD54 antibody with potent anti-myeloma activity developed by cellular panning of phage display libraries. Oncotarget 2017, 8, 77552–77566. [Google Scholar] [CrossRef] [Green Version]
- Klausz, K.; Cieker, M.; Kellner, C.; Rösner, T.; Otte, A.; Krohn, S.; Lux, A.; Nimmerjahn, F.; Valerius, T.; Gramatzki, M.; et al. Fc-engineering significantly improves the recruitment of immune effector cells by anti-ICAM-1 antibody MSH-TP15 for myeloma therapy. Haematology 2020. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Van Epps, H.; Anderson, M.; Yu, C.; Klussman, K.; Westendorf, L.; Carosino, C.; Manlove, L.; Cochran, J.; Neale, J.; Benjamin, D.; et al. Abstract 3833: SEA-BCMA: A highly active enhanced antibody for multiple myeloma. Immunology 2018, 78, 3833. [Google Scholar] [CrossRef]
- Li, F.; Ravetch, J.V. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proc. Natl. Acad. Sci. USA 2012, 109, 10966–10971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.L.; Dou, L.; Chan, H.T.C.; Field, V.L.; Mockridge, C.I.; Moss, K.; Williams, E.L.; Booth, S.G.; French, R.R.; Potter, E.A.; et al. Fcγ Receptor Dependency of Agonistic CD40 Antibody in Lymphoma Therapy Can Be Overcome through Antibody Multimerization. J. Immunol. 2014, 193, 1828–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overdijk, M.B.; Strumane, K.; Beurskens, F.J.; Buijsse, A.O.; Vermot-Desroches, C.; Vuillermoz, B.S.; Kroes, T.; De Jong, B.; Hoevenaars, N.; Hibbert, R.G.; et al. Dual epitope targeting and enhanced hexamerization by DR5 antibodies as a novel approach to induce potent anti-tumor activity through DR5 agonism. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Fc-Engineered mAb | Target | Iso-Type | Chimeric/Human(ized) | Fc Engineering Strategy | Enhanced Effector Function | Additional mAb Engineering | Clinical Stage; NCT of Recruiting Clinical Trials | Major Indication(s) |
|---|---|---|---|---|---|---|---|---|
| Obinutuzumab (GA101;Gazyva) | CD20, type II | IgG2 | Humanized | Afucosylation | ADCC | Modified elbow hinge | FDA-approved | FL and CLL |
| Ublituximab (LFB-R603, EMAB-6) | CD20, type I | IgG1 | Chimeric | Low fucose | ADCC | Phase 2/3 1 | CLL and B-NHL | |
| Ocaratuzumab (AME-133v, LY2469298) | CD20, type I | IgG1 | Humanized | Mutations P247I/A339Q | ADCC | Antigen binding affinity optimized | Discontinued | |
| PRO131921 (RhuMAb; v114) | CD20, type I | IgG1 | Humanized | Mutations (na) | ADCC and CDC | Discontinued | ||
| Ocrelizumab | CD20 | IgG1 | Humanized | Mutations (na) | ADCC | Discontinued in hematology, approved for MS | MS | |
| CD20 double engineered | CD20 | IgG1 | Afucosylation+ mutations S267E/H268F/S324T/G236A/I332E | ADCC and CDC | Preclinical | |||
| CD20 | IgG1/IgG3 | Afucosylation + mixed IgG1/IgG3 isotype | ADCC and CDC | Preclinical | ||||
| BI 836826 | CD37 | IgG1 | Chimeric | Mutations S239D/I332E | ADCC | Discontinued | ||
| DuoHexaBody-CD37 (GEN3009) | CD37 | IgG1 | Human | Mutation E430G (HexaBody) | CDC | Dual-epitope targeting | Phase 1 2 | B-NHL |
| Ianalumab (VAY736; B-1239) | BAFF-R | IgG1 | Human | Afucosylation | ADCC | Phase 1 3 | CLL | |
| Inebilizumab (MEDI-551) | CD19 | IgG1κ | Humanized | Afucosylation | ADCC | Phase 1/2 | B-cell malignancies | |
| MDX-1342 | CD19 | Human | Afucosylation | ADCC and ADCP | Phase 1 halted | |||
| Tafasitamab (MOR208, XmAb5574) | CD19 | IgG1 | Humanized | Mutations S239D/I332E | ADCC and ADCP | Antigen binding affinity optimized | Priority review granted by FDA | CLL and DLBCL |
| HexaBody-CD38 (GEN3014) | CD38 | IgG1 | Human | Mutation E430G (HexaBody) | CDC | Preclinical | B-NHL and MM | |
| anti-CD38 SIFbody | CD38 | Fc multimerization | ADCC and CDC | Preclinical | MM | |||
| XmAb5592 | HM1.24 | IgG1 | Humanized | Mutations S239D/I332E | MM | |||
| ICAM-1 | IgG1 | Human | Mutations S239D/I332E | ADCC and ADCP | Preclinical | MM | ||
| SEA-BCMA | BCMA | IgG1 | Humanized | Afucosylation | ADCC and ADCP | Phase 1 4 | MM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Horst, H.J.; Nijhof, I.S.; Mutis, T.; Chamuleau, M.E.D. Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers 2020, 12, 3041. https://doi.org/10.3390/cancers12103041
van der Horst HJ, Nijhof IS, Mutis T, Chamuleau MED. Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers. 2020; 12(10):3041. https://doi.org/10.3390/cancers12103041
Chicago/Turabian Stylevan der Horst, Hilma J., Inger S. Nijhof, Tuna Mutis, and Martine E. D. Chamuleau. 2020. "Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies" Cancers 12, no. 10: 3041. https://doi.org/10.3390/cancers12103041
APA Stylevan der Horst, H. J., Nijhof, I. S., Mutis, T., & Chamuleau, M. E. D. (2020). Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers, 12(10), 3041. https://doi.org/10.3390/cancers12103041