Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models

 , , ,
, , , 
 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mitochondrial Differences in Sorafenib vs. Regorafenib Experimental Liver Cancer Treatment
2.2. BCL-xL Antagonism Is Effective to Potentiate Regorafenib Activity Against Liver Cancer Cells
2.3. A-1331852 Addition to Regorafenib-Treated Hepatoma Cells Triggers MMP Loss and Mitochondrial-Mediated Caspase-Dependent Apoptotic Cell Death
2.4. Regorafenib Reduction of MCL-1 Facilitates A-1331852 Induction of Cell Death in Liver Cancer Cells
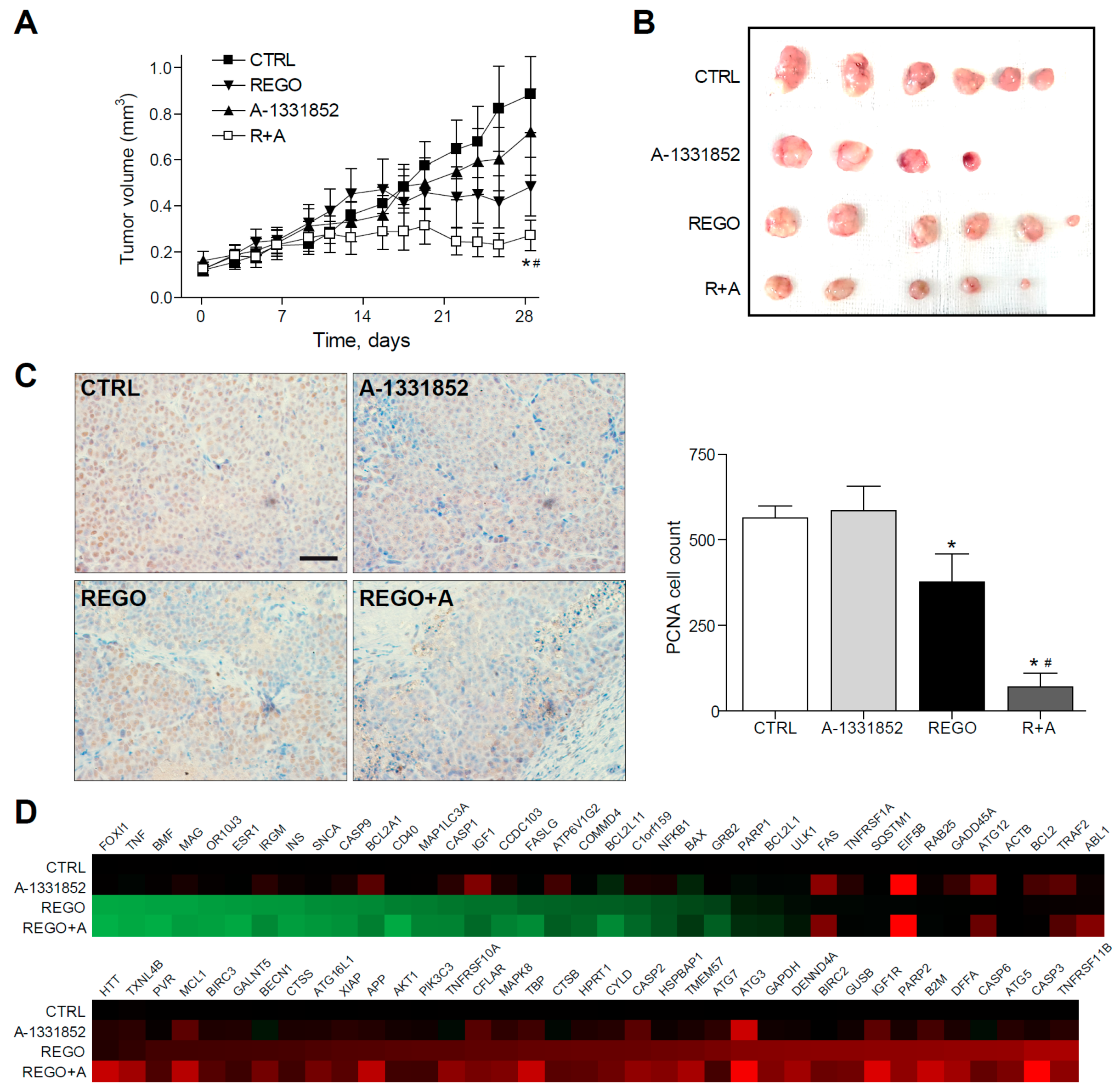
2.5. A-1331852 in Combination with Regorafenib Is Effective to Reduce Liver Cancer Progression in a PDX Mouse Model
2.6. Regorafenib Resistant Cells Are Sensitive to A-1331852 Co-Administration in Vitro and In Vivo
2.7. BCL-xL Upregulation and MCL-1 Reduction Are Present in HCC Tumor Tissue
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Biochemical Analysis
4.3. MTT Assay
4.4. Crystal Violet Staining
4.5. Caspase-3 Activity Assay
4.6. Hoechst Staining
4.7. Mitochondrial Membrane Potential Assay
4.8. 3D Tumor Liver Spheroids Generation
4.9. Immunoblot Analysis
4.10. RNA Isolation and Real Time RT-PCR
- human BCL-2: Fw 5′-GGAGGATTGTGGCCTTCTTT-3′; Rv 5′-GCCGTACAGTTCCACAAAGG-3′ human BCL-xL: Fw 5′-GGATGGCCACTTACCTGA-3′; Rv 5′-CGGTTGAAGCGTTCCTG-3′
- human MCL-1: Fw 5′-ATGCTTCGGAAACTGGACAT-3′; Rv 5′-TCCTGATGCCACCTTCTAGG-3′ human ′-Act: Fw 5′-AGAAAATCTGGCACCACACC-3′ Rv 5′-AGAGGCGTACAGGGATAGCA-3′
4.11. Immunohistochemical Staining
4.12. Tumor Animal Models
4.13. Gene Array
4.14. cDNA Array
4.15. HCC Patient Study and ATLAS Database Information
4.16. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bruix, J.; Reig, M.; Sherman, M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Aspord, C.; Decaens, T. Predictive factors for response to PD-1/PD-L1 checkpoint inhibition in the field of hepatocellular carcinoma: Current status and challenges. Cancers 2019, 11, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wörns, M.-A.; Galle, P.R. HCC therapies—lessons learned. Nat. Revi. Gastroenterol. Hepatol. 2014, 11, 447–452. [Google Scholar] [CrossRef]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers (Basel) 2019, 11, 916. [Google Scholar] [CrossRef] [Green Version]
- Bhola Patrick, D.; Letai, A. Mitochondria—Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Al-harbi, S.; Hill, B.T.; Mazumder, S.; Singh, K.; DeVecchio, J.; Choudhary, G.; Rybicki, L.A.; Kalaycio, M.; Maciejewski, J.P.; Houghton, J.A.; et al. An anti-apoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood 2011, 118, 3579–3590. [Google Scholar] [CrossRef] [Green Version]
- Touzeau, C.; Ryan, J.; Guerriero, J.; Moreau, P.; Chonghaile, T.N.; Le Gouill, S.; Richardson, P.; Anderson, K.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2015, 30, 761–764. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Fernando, J.; Sancho, P.; Fernández-Rodriguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Barcena, C.; de Gregorio, E.; Moutinho, C.; Barbero-Camps, E.; Villanueva, A.; Colell, A.; Mari, M.; et al. Targeting glucosylceramide synthase upregulation reverts sorafenib resistance in experimental hepatocellular carcinoma. Oncotarget 2016, 7, 8253–8267. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, A.; Ezzoukhry, Z.; Francois, C.; Louandre, C.; Sabbagh, C.; Nguyen-Khac, E.; Descamps, V.; Trouillet, N.; Godin, C.; Regimbeau, J.M.; et al. BAD, a Proapoptotic Member of the BCL2 Family, Is a Potential Therapeutic Target in Hepatocellular Carcinoma. Mol. Cancer Res. 2010, 8, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Chiou, J.-F.; Tai, C.-J.; Wang, Y.-H.; Liu, T.-Z.; Jen, Y.-M.; Shiau, C.-Y. Sorafenib induces preferential apoptotic killing of a drug- and radio-resistant hep G2 cells through a mitochondria-dependent oxidative stress mechanism. Cancer Biol. Ther. 2009, 8, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Anti-apoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Du, Y.; Ma, H.; Liang, Q.; Zhu, X.; Tian, J. Preclinical comparison of regorafenib and sorafenib efficacy for hepatocellular carcinoma using multimodality molecular imaging. Cancer Lett. 2019, 453, 74–83. [Google Scholar] [CrossRef]
- Kissel, M.; Berndt, S.; Fiebig, L.; Kling, S.; Ji, Q.; Gu, Q.; Lang, T.; Hafner, F.T.; Teufel, M.; Zopf, D. Anti-tumor effects of regorafenib and sorafenib in preclinical models of hepatocellular carcinoma. Oncotarget 2017, 8, 107096–107108. [Google Scholar] [CrossRef]
- Butterworth, M.; Pettitt, A.; Varadarajan, S.; Cohen, G.M. BH3 profiling and a toolkit of BH3-mimetic drugs predict anti-apoptotic dependence of cancer cells. Br. J. Cancer 2016, 114, 638–641. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Bougie, P.; Maiga, S.; Tessoulin, B.; Bourcier, J.; Bonnet, A.; Rodriguez, M.S.; Le Gouill, S.; Touzeau, C.; Moreau, P.; Pellat-Deceunynck, C.; et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood 2018, 132, 2656–2669. [Google Scholar] [CrossRef] [Green Version]
- Villalobos-Ortiz, M.; Ryan, J.; Mashaka, T.N.; Opferman, J.T.; Letai, A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2019. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves anti-tumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Tan, S.; Zou, F.; Yu, J.; Zhang, L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017, 36, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Huang, F.; Yao, Z.; Jia, C.; Xiong, Z.; Liang, H.; Lin, N.; Deng, M. Inhibition of cyclin E1 sensitizes hepatocellular carcinoma cells to regorafenib by mcl-1 suppression. Cell Commun. Signal. 2019, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Tovar, V.; Alsinet, C.; Villanueva, A.; Hoshida, Y.; Chiang, D.Y.; Solé, M.; Thung, S.; Moyano, S.; Toffanin, S.; Mínguez, B.; et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 2010, 52, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Arai, H.; Battaglin, F.; Wang, J.; Lo, J.H.; Soni, S.; Zhang, W.; Lenz, H.J. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 2019, 81, 101912. [Google Scholar] [CrossRef]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; González-Gallego, J.; Mauriz, J.L. Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp. Mol. Med. 2019, 51, 109. [Google Scholar] [CrossRef] [Green Version]
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Lim, A.; Kim, R. Immunotherapy in Hepatocellular Carcinoma: Is There a Light at the End of the Tunnel? Cancers (Basel) 2019, 11, 1078. [Google Scholar] [CrossRef] [Green Version]
- Natri, H.M.; Wilson, M.A.; Buetow, K.H. Distinct molecular etiologies of male and female hepatocellular carcinoma. BMC Cancer 2019, 19, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faqar-Uz-Zaman, S.F.; Heinicke, U.; Meister, M.T.; Vogler, M.; Fulda, S. BCL-xL-selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Lett. 2018, 412, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Rello-Varona, S.; Fuentes-Guirado, M.; López-Alemany, R.; Contreras-Pérez, A.; Mulet-Margalef, N.; García-Monclús, S.; Tirado, O.M.; Del Muro, X.G. Bcl-xL inhibition enhances Dinaciclib-induced cell death in soft-tissue sarcomas. Sci. Rep. 2019, 9, 3816. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncsek, A.; Al-Suraih, M.S.; Trussoni, C.E.; O’Hara, S.P.; Splinter, P.L.; Zuber, C.; Patsenker, E.; Valli, P.V.; Fingas, C.D.; Weber, A.; et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 2018, 67, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent anti-tumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- May, J.E.; Morse, H.R.; Xu, J.; Donaldson, C. Development of a novel, physiologically relevant cytotoxicity model: Application to the study of chemotherapeutic damage to mesenchymal stromal cells. Toxicol. Appl. Pharmacol. 2012, 263, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Van Tienderen, G.S.; Groot Koerkamp, B.; IJzermans, J.N.M.; van der Laan, L.J.W.; Verstegen, M.M.A. Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment. Cancers (Basel) 2019, 11, 1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Martinez, L.; García-Ruiz, C.; de Mingo, A.; Caballeria, J.; Fernandez-Checa, J.C.; Marí, M.; et al. Angiogenin secretion from hepatoma cells activates hepatic stellate cells to amplify a self-sustained cycle promoting liver cancer. Sci. Rep. 2015, 5, 7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mingo, Á.; de Gregorio, E.; Moles, A.; Tarrats, N.; Tutusaus, A.; Colell, A.; Fernandez-Checa, J.C.; Morales, A.; Marí, M. Cysteine cathepsins control hepatic NF-κB-dependent inflammation via sirtuin-1 regulation. Cell Death Dis. 2016, 7, e2464. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucarull, B.; Tutusaus, A.; Subías, M.; Stefanovic, M.; Hernáez-Alsina, T.; Boix, L.; Reig, M.; García de Frutos, P.; Marí, M.; Colell, A.; et al. Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models. Cancers 2020, 12, 332. https://doi.org/10.3390/cancers12020332
Cucarull B, Tutusaus A, Subías M, Stefanovic M, Hernáez-Alsina T, Boix L, Reig M, García de Frutos P, Marí M, Colell A, et al. Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models. Cancers. 2020; 12(2):332. https://doi.org/10.3390/cancers12020332
Chicago/Turabian StyleCucarull, Blanca, Anna Tutusaus, Miguel Subías, Milica Stefanovic, Tania Hernáez-Alsina, Loreto Boix, María Reig, Pablo García de Frutos, Montserrat Marí, Anna Colell, and et al. 2020. "Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models" Cancers 12, no. 2: 332. https://doi.org/10.3390/cancers12020332
APA StyleCucarull, B., Tutusaus, A., Subías, M., Stefanovic, M., Hernáez-Alsina, T., Boix, L., Reig, M., García de Frutos, P., Marí, M., Colell, A., Bruix, J., & Morales, A. (2020). Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models. Cancers, 12(2), 332. https://doi.org/10.3390/cancers12020332