High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Patients
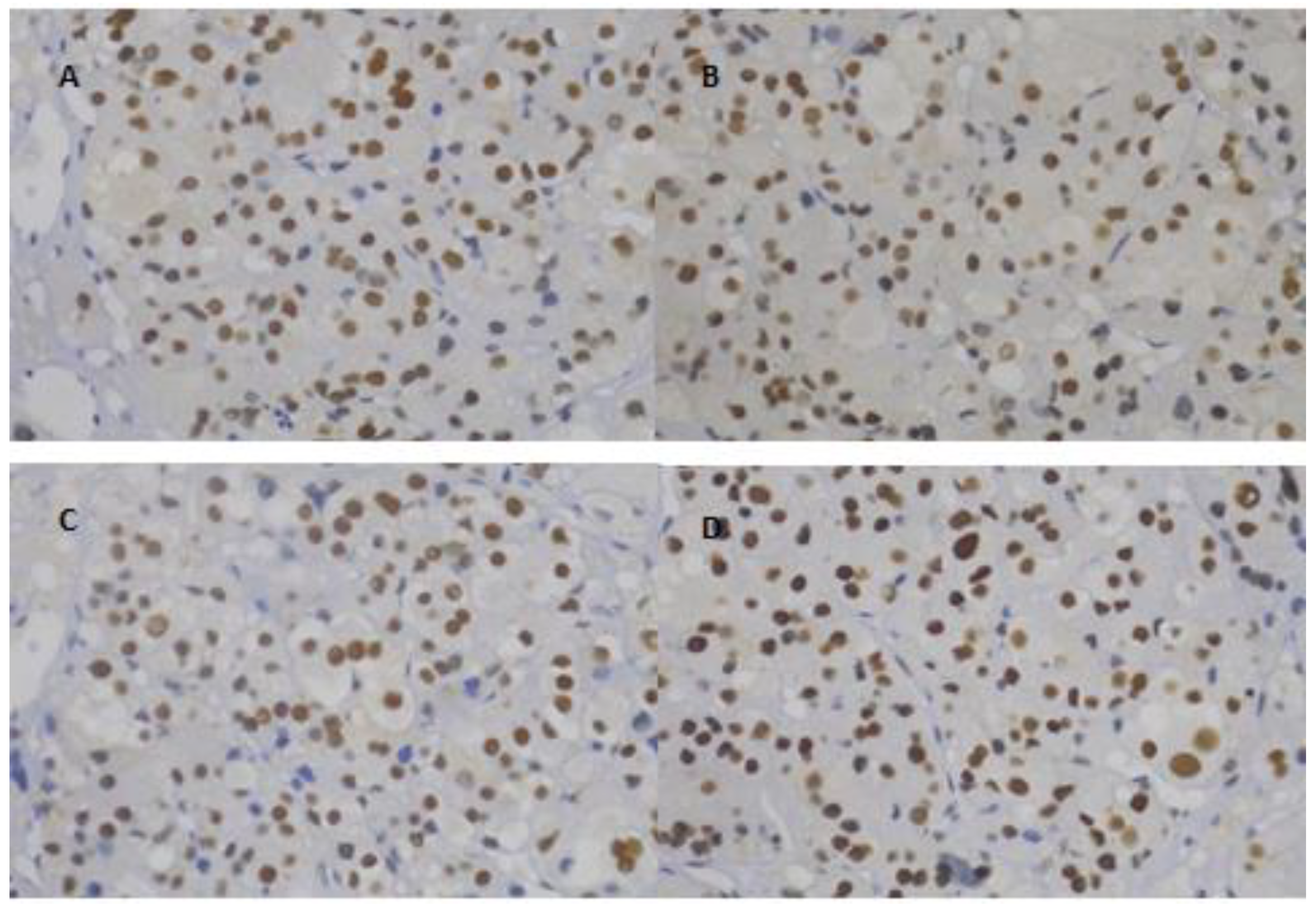
2.2. Immunohistochemistry
2.3. Genetic Analysis
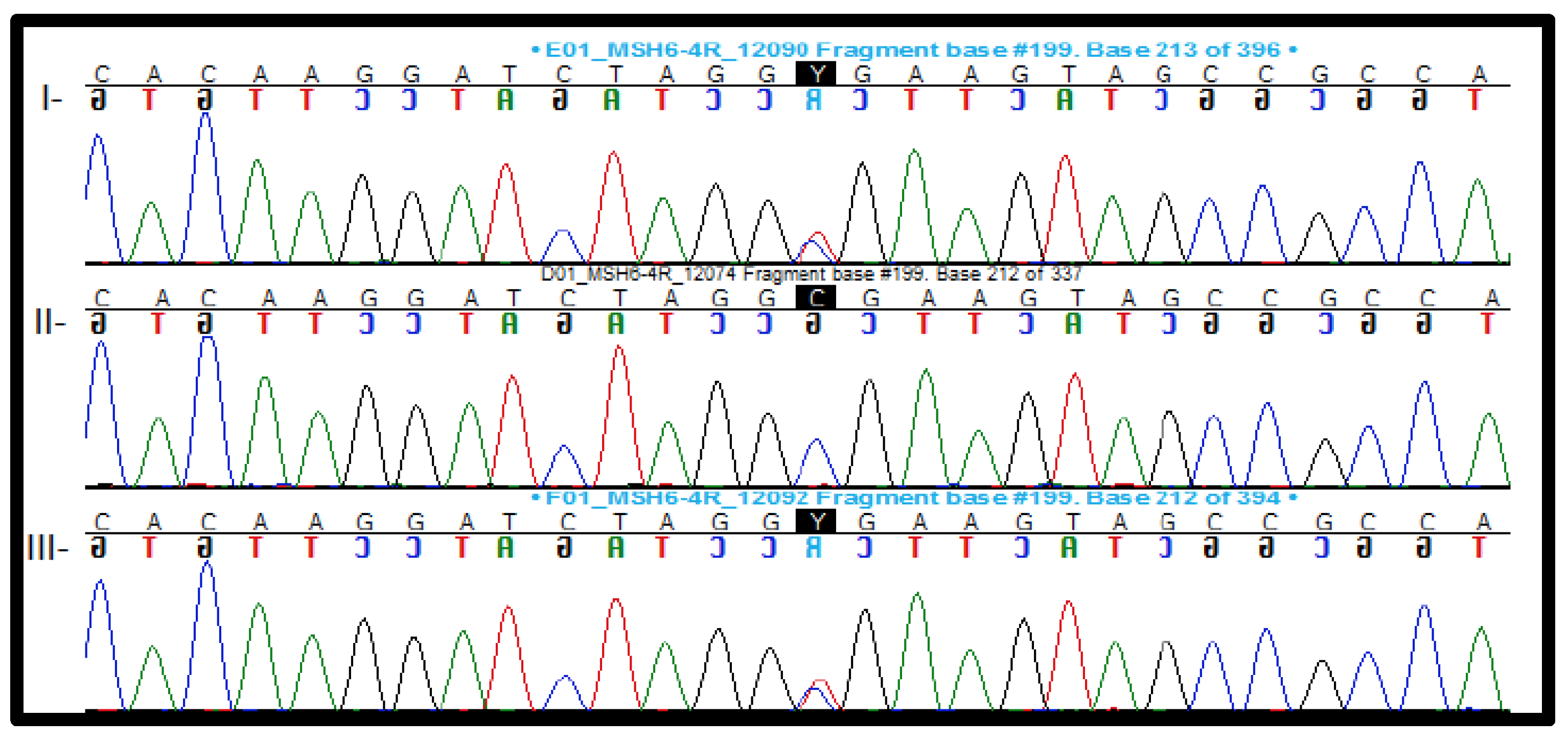
2.3.1. Pathogenic and Likely Pathogenic Variants
2.3.2. Variant of Uncertain Significance (VUS)
2.3.3. Copy Number Variation
2.4. Family History of Lynch Syndrome-Associated Tumors
3. Discussion
4. Materials and Methods
4.1. Immunohistochemistry
4.1.1. Tissue Microarray (TMA) and Immunohistochemical Analysis of the TMA
4.1.2. Formalin-Fixed and Paraffin-Embedded Tumor Sections
4.2. Genetic Analysis
4.2.1. Panel Sequencing and Data Analysis
4.2.2. Sanger Sequencing
4.2.3. MLPA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bilimoria, K.Y.; Shen, W.T.; Elaraj, D. Adrenocortical Carcinoma in the United States: Treatment utilization and prognostic factors. Cancer 2008, 113, 3130–3136. [Google Scholar] [CrossRef]
- Dackiw, A.P.; Lee, J.E.; Gagel, R.F.; Evans, D.B. Adrenal cortical carcinoma. World J. Surg. 2001, 25, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Reiff, E.; Duh, Q.Y.; Clark, O.H.; McMillan, A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress? World J. Surg. 2006, 30, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Dekkers, O.M.; Else, T. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A. Adrenocortical Carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrini, R.; Ribeiro, R.C.; DeLacerda, L. Childhood adrenocortical tumors. J. Clin. Endocrinol. Metab. 1997, 82, 2027–2031. [Google Scholar] [CrossRef]
- Wajchenberg, B.L.; Albergaria Pereira, M.A.; Mendonca, B.B. Adrenocortical carcinoma-clinical and laboratory observations. Cancer 2000, 88, 711–736. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Fragoso, M.C.; Lotfi, C.F. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J. Clin. Endocrinol. Metab. 2008, 93, 3524–3531. [Google Scholar] [CrossRef] [Green Version]
- Pinto, E.M.; Billerbeck, A.E.; Villares, M.C.; Domenice, S.; Mendonca, B.B.; Latronico, A.C. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq. Bras. Endocrinol. Metabol. 2004, 48, 647–650. [Google Scholar] [CrossRef] [Green Version]
- Latronico, A.C.; Pinto, E.M.; Domenice, S. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4970–4973. [Google Scholar] [CrossRef]
- Custodio, G.; Komechen, H.; Figueiredo, F.R.; Fachin, N.D.; Pianovski, M.A.; Figueiredo, B.C. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol. Cell. Endocrinol. 2012, 351, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B. A inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.P.; Fraumeni, J.F., Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M. Epidemiology of adrenocortical carcinoma. In Adrenocortical Carcinoma, 1st ed.; Hammer, G., Else, T., Eds.; Springer: New York, NY, USA, 2009; pp. 23–29. [Google Scholar]
- Lynch, H.T.; Shaw, M.W.; Magnuson, C.W.; Larsen, A.L.; Krush, A.J. Hereditary Factors In Cancer. Study of two large midwestern kindreds. Arch. Intern. Med. 1966, 117, 206–212. [Google Scholar] [CrossRef]
- Vasen, H.F.A.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch Syndrome) proposed by the International Collaborative Group n HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F. Mutation of a mutL homolog in hereditary colo cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T. Mutation in the DNA mismatch repair gene homolog hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.E.; Papp, J.; Szentirmay, Z.; Otto, S.; Olah, E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum. Mutat. 2009, 30, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Raymond, V.M.; Everett, J.N.; Furtado, L.V. Adrenocortical carcinoma is a Lynch syndrome-associated cancer. J. Clin. Oncol. 2013, 31, 3012–3018. [Google Scholar] [CrossRef]
- Mandelker, D.; Dal Cin, P.; Jacene, H.A.; Armand, P.; Stone, R.M.; Lindeman, N.I. Refractory myeloid sarcoma with a FIP1L1-PDGFRA rearrangement detected by clinical high throughput somatic sequencing. Exp. Hematol. Oncol. 2015, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Wielandt, A.M.; Zarate, A.J.; Hurtado, C. Lynch syndrome: Selection of families by microsatellite instability and immunohistochemistry. Rev. Med. Chile 2012, 140, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Chang, M.; Chang, H.M.; Chang, F. Microsatellite Instability: A Predictive Biomarker for Cancer Immunotherapy. Appl. Immunohistochem. Mol. Morphol. 2018, 26, e15–e21. [Google Scholar] [CrossRef]
- Caron, O. Endometrial cancers and predisposition. Bull. Cancer 2012, 99, 99–105. [Google Scholar] [CrossRef]
- Palomaki, G.E.; McClain, M.R.; Melillo, S.; Hampel, H.L.; Thibodeau, S.N. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet. Med. 2009, 11, 42–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Ries, L.A.G.; Melbert, D.; Krapcho, M.; Stinchcomb, D.G.; Howlader, N.; Horner, M.J.; Mariotto, A.; Miller, B.A.; Feuer, E.J.; Altekruse, S.F.; et al. SEER Cancer Statistics Review, 1975–2005; National Cancer Institute: Bethesda, MD, USA, 2008.
- Huang, S.C.; Durno, C.A.; Erdman, S.H. Lynch syndrome: A pediatric perspective. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Vasen, H.F.; Boland, C.R. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA 2005, 293, 2028–2030. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Dillon, J.L.; Gonzalez, J.L.; DeMars, L.; Bloch, K.J.; Tafe, L.J. Universal screening for Lynch syndrome in endometrial cancers: Frequency of germline mutations and identification of patients with Lynch-like syndrome. Hum Pathol. 2017, 70, 121–128. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Capo-Chichi, J.M.; Spence, T. Heterogenous loss of mismatch repair (MMR) protein expression: A challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J. Pathol. Clin. Res. 2019, 5, 115–129. [Google Scholar] [CrossRef]
- Jones, J.S.; Chi, X.; Gu, X.; Lynch, P.M.; Amos, C.I.; Frazier, M.L. p53 polymorphism and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Clin. Cancer Res. 2004, 10, 5845–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.R.; Liu-Mares, W.; Van Emburgh, B.O. Genetic polymorphisms of multiple DNA repair pathways impact age at diagnosis and TP53 mutations in breast cancer. Carcinogenesis 2011, 32, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; Keuling, A.M.; Lai, R.; Nation, P.N.; Tron, V.A.; Andrew, S.E. The associated contributions of p53 and the DNA mismatch repair protein Msh6 to spontaneous tumorigenesis. Carcinogenesis 2007, 28, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.A.; Reece, J.C.; Buchanan, D.D. Childhood cancers in families with and without Lynch syndrome. Fam. Cancer 2015, 14, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, C.P.; Achatz, M.I.; Brugieres, L. Cancer screening recommendations for individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lupat, R.; Amarasinghe, K.C. CONTRA: Copy number analysis for targeted resequencing. Bioinformatics 2012, 28, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patients | Gender | Age (yrs) | Clinical Presentation | Metastasis | Global Survival Time (yrs) | MMR Genes Variants | InSiGHT DNA Variant Database | ACMG Classification | |
|---|---|---|---|---|---|---|---|---|---|
| #6 | M | 2.2 | V | No | 7.54 | MLH1-c.1500_1502del p.Ile501del | Class 3: uncertain | Likely pathogenic | PM1; PM2;PM4; PP3 |
| #9 | M | 2.1 | V | No | 8.19 | MLH1-c.1808C > T p.Pro603Leu | Not identified | Likely pathogenic | PM1, PM2, PP2, PP3 |
| #16 | F | 3.3 | V/C | Yes | 8.07 | MSH6-c.2420A > G p.Tyr807Cys | Not identified | VUS | PM2, PP3, BP1 |
| #21 | F | 3 | V/C | No | 31.61 | MSH6-c.359T > C p.Ile120Thr | Not identified | Likely benign | PM1, PM2, BP3, BP4 |
| #33 | M | 11.5 | V/C | No | 3.14 | MSH6-c.328C > T p.Arg110X | Not identified | Pathogenic | PVS1, PM1, PM2, PP3, PP5 |
| #35 | M | 2.3 | V | No | 0.08 | MSH6-c.643G > T p.Val215Leu | Not identified | Likely benign | PM1, PM2, BP3, BP4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brondani, V.B.; Montenegro, L.; Lacombe, A.M.F.; Magalhães, B.M.; Nishi, M.Y.; Funari, M.F.d.A.; Narcizo, A.d.M.; Cardoso, L.C.; Siqueira, S.A.C.; Zerbini, M.C.N.; et al. High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers 2020, 12, 621. https://doi.org/10.3390/cancers12030621
Brondani VB, Montenegro L, Lacombe AMF, Magalhães BM, Nishi MY, Funari MFdA, Narcizo AdM, Cardoso LC, Siqueira SAC, Zerbini MCN, et al. High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers. 2020; 12(3):621. https://doi.org/10.3390/cancers12030621
Chicago/Turabian StyleBrondani, Vania Balderrama, Luciana Montenegro, Amanda Meneses Ferreira Lacombe, Breno Marchiori Magalhães, Mirian Yumie Nishi, Mariana Ferreira de Assis Funari, Amanda de Moraes Narcizo, Lais Cavalca Cardoso, Sheila Aparecida Coelho Siqueira, Maria Claudia Nogueira Zerbini, and et al. 2020. "High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53" Cancers 12, no. 3: 621. https://doi.org/10.3390/cancers12030621
APA StyleBrondani, V. B., Montenegro, L., Lacombe, A. M. F., Magalhães, B. M., Nishi, M. Y., Funari, M. F. d. A., Narcizo, A. d. M., Cardoso, L. C., Siqueira, S. A. C., Zerbini, M. C. N., Denes, F. T., Latronico, A. C., Mendonca, B. B., Almeida, M. Q., Lerario, A. M., Soares, I. C., & Fragoso, M. C. B. V. (2020). High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers, 12(3), 621. https://doi.org/10.3390/cancers12030621