Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy

 ,
,
Abstract
:1. Introduction
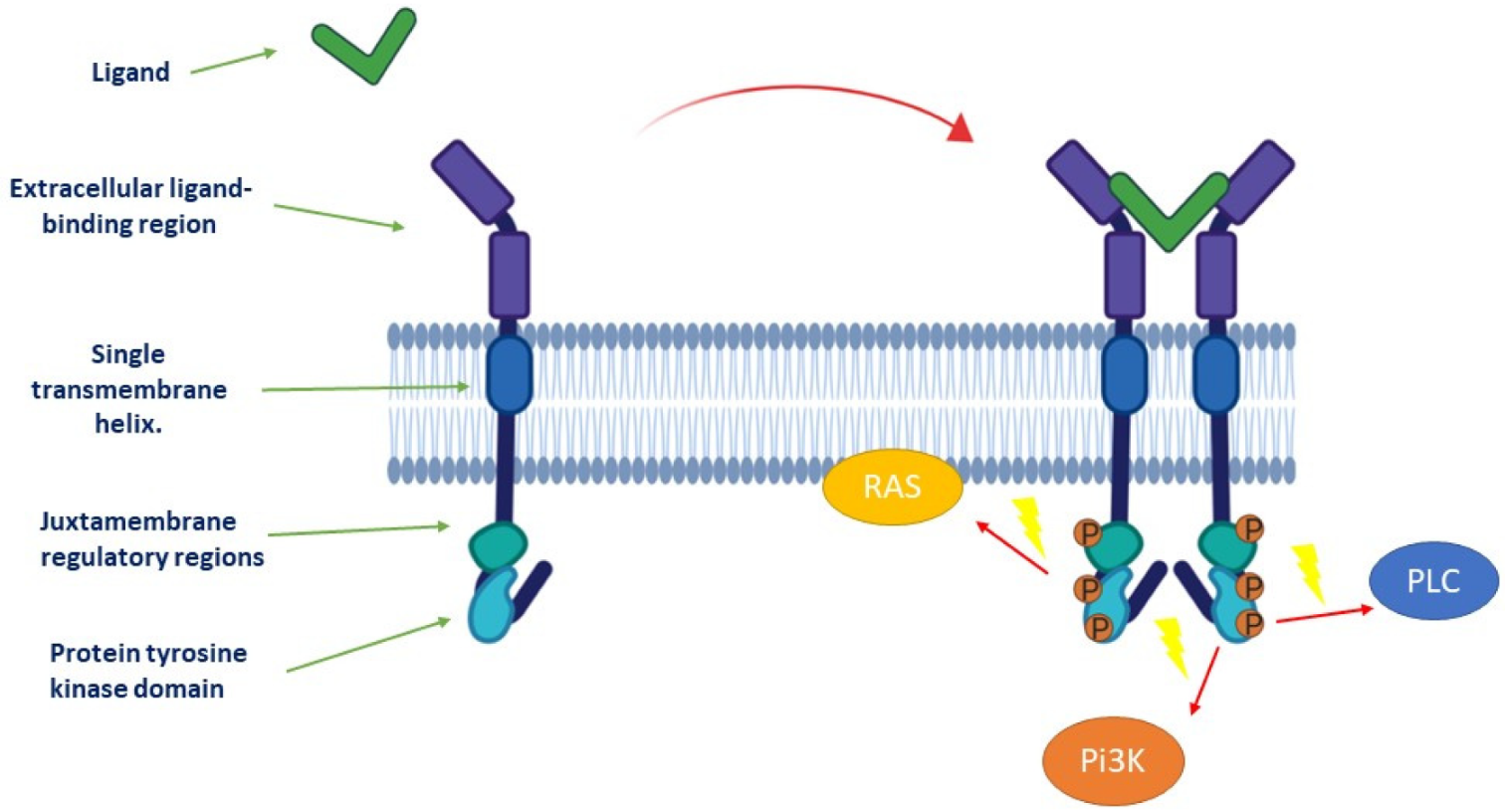
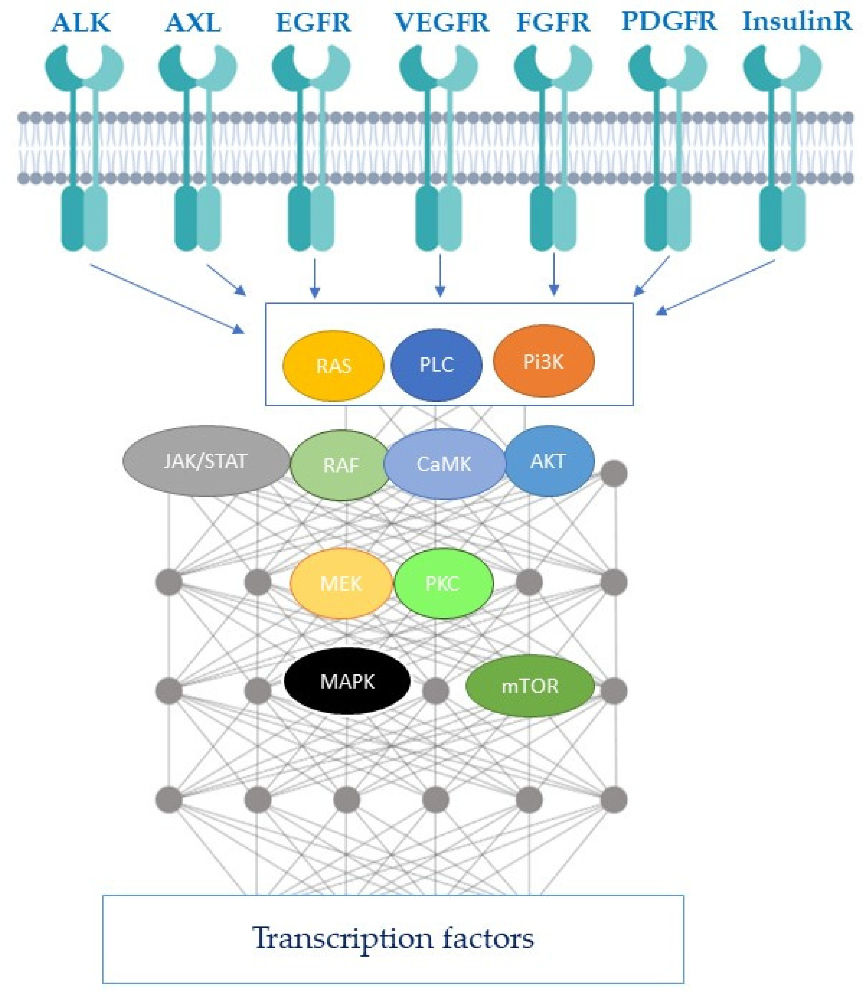
1.1. Description of Receptor Tyrosine Kinases and Downstream Signaling Pathways
1.2. Classification of Receptor Tyrosine Kinase Inhibitors
2. Current Place of Receptor Tyrosine Kinase Inhibitors in Oncological Treatments
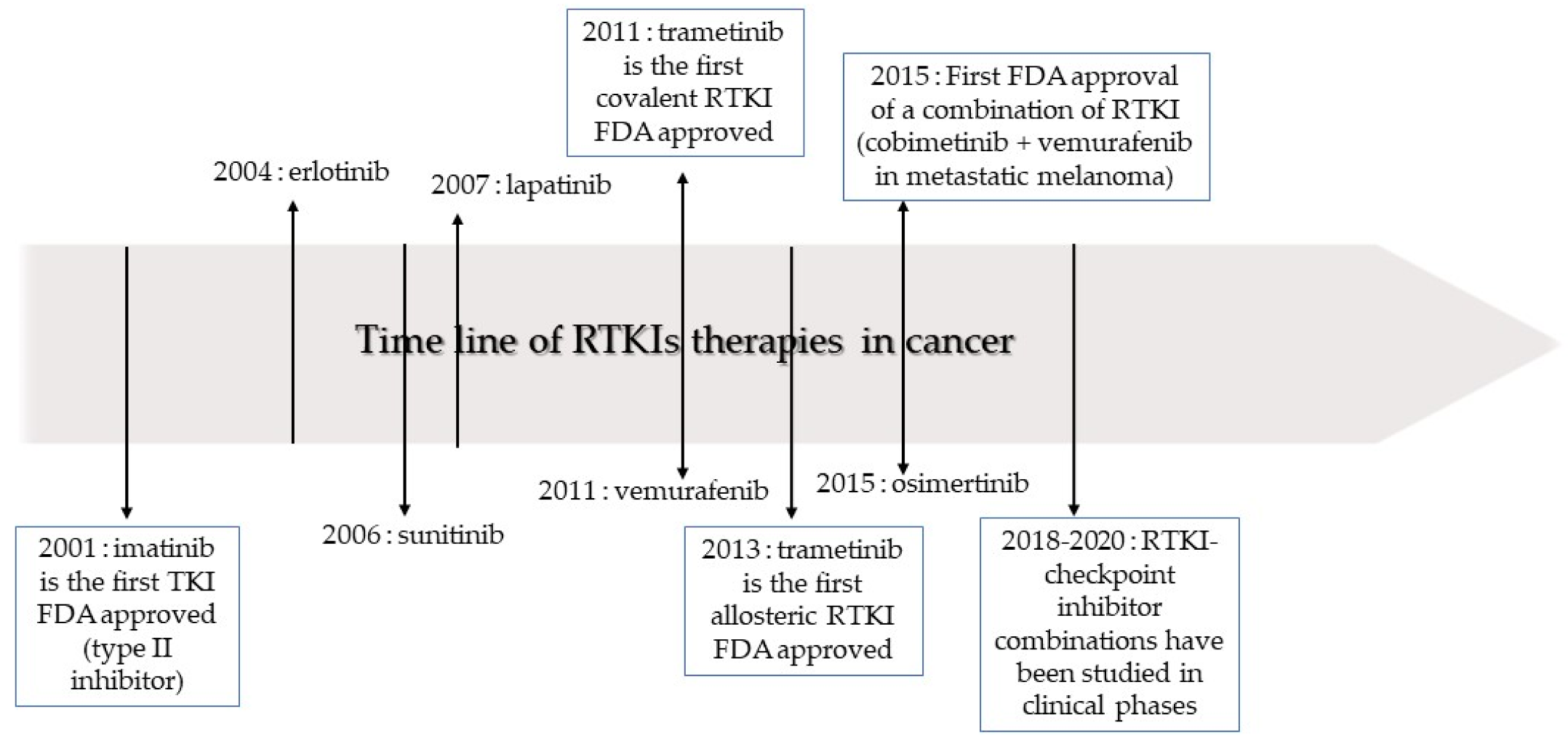
2.1. Evolution of Receptor Tyrosine Kinase Inhibitor Use in Non-Small Cell Lung Cancer
2.2. Current Indications of Receptor Tyrosine Kinase Inhibitors in Other Types of Cancer
2.3. Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibitors
3. Improving the Use of Receptor Tyrosine Kinase Inhibitors: Combinatorial Treatment without Increasing Toxicity
3.1. Receptor Tyrosine Kinase Inhibitor Combinations
3.2. Receptor Tyrosine Kinase Inhibitors and Synthetic Lethality
3.3. Impact of the Non-Immune Microenvironment on Receptor Tyrosine Kinase Inhibitor Efficacy
3.4. Impact of the Immune Microenvironment on Receptor Tyrosine Kinase Inhibitor Efficacy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Liebmann, C. Regulation of MAP kinase activity by peptide receptor signalling pathway: Paradigms of multiplicity. Cell Signal. 2001, 13, 777–785. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 184–187. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisci, S.; Amitrano, F.; Saggese, M.; Muto, T.; Sarno, S.; Mele, S.; Vitale, P.; Ronga, G.; Berretta, M.; Di Francia, R. Overview of Current Targeted Anti-Cancer Drugs for Therapy in Onco-Hematology. Medicina 2019, 55, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Panicker, R.C.; Chattopadhaya, S.; Coyne, A.G.; Srinivasan, R. Allosteric Small-Molecule Serine/Threonine Kinase Inhibitors. Adv. Exp. Med. Biol. 2019, 1163, 253–278. [Google Scholar] [CrossRef]
- Pacenta, H.L.; Macy, M.E. Entrectinib and other ALK/TRK inhibitors for the treatment of neuroblastoma. Drug Des. Dev. Ther. 2018, 12, 3549–3561. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.; Youn, C.; Moon, A.R.; Howland, A.; Armstrong, C.A.; Song, P.I. Therapeutic Inhibitors against Mutated BRAF and MEK for the Treatment of Metastatic Melanoma. Chonnam. Med. J. 2017, 53, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Hochmair, M.J.; Morabito, A.; Hao, D.; Yang, C.T.; Soo, R.A.; Yang, J.C.; Gucalp, R.; Halmos, B.; Wang, L.; Marten, A.; et al. Sequential afatinib and osimertinib in patients with EGFR mutation-positive non-small-cell lung cancer: Updated analysis of the observational GioTag study. Future Oncol. 2019, 15, 2905–2914. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Correction to: “Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann. Oncol. 2019, 30, 863–870. [Google Scholar] [CrossRef]
- Pennell, N.A.; Lynch, T.J., Jr. Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist 2009, 14, 399–411. [Google Scholar] [CrossRef]
- NIH. Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT03122717 (accessed on 17 March 2020).
- Zhang, N.; Liang, C.; Song, W.; Tao, D.; Yao, J.; Wang, S.; Ma, L.; Shi, Y.; Han, X. Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. J. Cancer 2019, 10, 1275–1287. [Google Scholar] [CrossRef]
- Yang, Z.; Tam, K.Y. Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grunwald, V.; Gillessen, S.; Horwich, A.; Committee, E.G. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, A.; Miyake, H.; Fujisawa, M. Molecular mechanism mediating cytotoxic activity of axitinib in sunitinib-resistant human renal cell carcinoma cells. Clin. Transl. Oncol. 2016, 18, 893–900. [Google Scholar] [CrossRef]
- Yu, S.S.; Quinn, D.I.; Dorff, T.B. Clinical use of cabozantinib in the treatment of advanced kidney cancer: Efficacy, safety, and patient selection. Onco Targets Ther. 2016, 9, 5825–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; Andre, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)dagger. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A.; Committee, E.G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1856–1883. [Google Scholar] [CrossRef] [Green Version]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; Committee, E.G. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Horne, S.D.; Stevens, J.B.; Abdallah, B.Y.; Liu, G.; Bremer, S.W.; Ye, C.J.; Heng, H.H. Why imatinib remains an exception of cancer research. J. Cell Physiol. 2013, 228, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Saussele, S.; Rosti, G.; Mahon, F.X.; Janssen, J.; Hjorth-Hansen, H.; Richter, J.; Buske, C.; Committee, E.G. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv41–iv51. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [Green Version]
- Foster, R.; Griffith, R.; Ferrao, P.; Ashman, L. Molecular basis of the constitutive activity and STI571 resistance of Asp816Val mutant KIT receptor tyrosine kinase. J. Mol. Graph. Model. 2004, 23, 139–152. [Google Scholar] [CrossRef]
- Roberts, K.G.; Odell, A.F.; Byrnes, E.M.; Baleato, R.M.; Griffith, R.; Lyons, A.B.; Ashman, L.K. Resistance to c-KIT kinase inhibitors conferred by V654A mutation. Mol. Cancer Ther. 2007, 6, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Alexander, P.B.; Wang, X.F. Resistance to receptor tyrosine kinase inhibition in cancer: Molecular mechanisms and therapeutic strategies. Front. Med. 2015, 9, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Del Re, M.; Rofi, E.; Restante, G.; Crucitta, S.; Arrigoni, E.; Fogli, S.; Di Maio, M.; Petrini, I.; Danesi, R. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget 2018, 9, 6630–6643. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Hauschild, A.; Lindenblatt, N.; Pentheroudakis, G.; Keilholz, U.; Committee, E.G. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v126–v132. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef]
- Moradpour, Z.; Barghi, L. Novel Approaches for Efficient Delivery of Tyrosine Kinase Inhibitors. J. Pharm. Pharm. Sci. 2019, 22, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Ding, Y.; Gong, C.; Huang, D.; Chen, R.; Sui, P.; Lin, K.H.; Liang, G.; Yuan, L.; Xiang, H.; Chen, J.; et al. Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat. Commun. 2018, 9, 4274. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Y.; Gong, P.; Gabrilove, J.; Waxman, S.; Jing, Y. Arsenic Trioxide and Sorafenib Induce Synthetic Lethality of FLT3-ITD Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1871–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottier, C.; Wheatherspoon, A.; Roncarati, P.; Longuespee, R.; Herfs, M.; Duray, A.; Delvenne, P.; Quatresooz, P. The importance of the tumor microenvironment in the therapeutic management of cancer. Expert Rev. Anticancer Ther. 2015, 15, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Prete, A.; Lo, A.S.; Sadow, P.M.; Bhasin, S.S.; Antonello, Z.A.; Vodopivec, D.M.; Ullas, S.; Sims, J.N.; Clohessy, J.; Dvorak, A.M.; et al. Pericytes Elicit Resistance to Vemurafenib and Sorafenib Therapy in Thyroid Carcinoma via the TSP-1/TGFbeta1 Axis. Clin. Cancer Res. 2018, 24, 6078–6097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.S.; Dane, M.; Chin, K.; Tatarova, Z.; Liu, M.; Liby, T.; Thompson, W.; Smith, R.; Nederlof, M.; Bucher, E.; et al. Microenvironment-Mediated Mechanisms of Resistance to HER2 Inhibitors Differ between HER2+ Breast Cancer Subtypes. Cell Syst. 2018, 6, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef] [Green Version]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Patnaik, A.; Swanson, K.D.; Csizmadia, E.; Solanki, A.; Landon-Brace, N.; Gehring, M.P.; Helenius, K.; Olson, B.M.; Pyzer, A.R.; Wang, L.C.; et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017, 7, 750–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, L.M.A.; Fernandez, I.P.; Cassinello, J. Tyrosine kinase inhibitors reprogramming immunity in renal cell carcinoma: Rethinking cancer immunotherapy. Clin. Transl. Oncol. 2017, 19, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Sawa, K.; Fukui, M.; Oyanagi, J.; Izumi, M.; Ogawa, K.; Suzumura, T.; Watanabe, T.; Kaneda, H.; Mitsuoka, S.; et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with EGFR-mutant non-small cell lung cancer. Cancer Sci. 2019, 110, 3244–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.H.; Lee, C.H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Hara, H.; Fukuoka, S.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib plus nivolumab in patients with advanced colorectal or gastric cancer: An open-label, dose-finding, and dose-expansion phase 1b trial (REGONIVO, EPOC1603). Ann. Oncol. 2019, 30 (Suppl. 4), iv124. [Google Scholar] [CrossRef]
- Akalu, Y.T.; Rothlin, C.V.; Ghosh, S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol. Rev. 2017, 276, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Durden, D.L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 5245034. [Google Scholar] [CrossRef] [Green Version]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y.C.; et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Bopaiah, J.; Alghamedy, F.; Jacobs, N.; Weiss, H.L.; de Jong, W.A.; Ellingson, S.R. Polypharmacology Within the Full Kinome: A Machine Learning Approach. AMIA Jt. Summits Transl. Sci. Proc. 2018, 2017, 98–107. [Google Scholar] [PubMed]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Dixon, K.; Farrant, E.; Feng, Q.; Gibson, K.R.; van Hoorn, W.P.; Mills, J.; Morgan, T.; Parry, D.M.; Ramjee, M.K.; et al. Rapid discovery of a novel series of Abl kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J. Med. Chem. 2013, 56, 3033–3047. [Google Scholar] [CrossRef] [PubMed]
- Benz, M.; Molla, M.R.; Boser, A.; Rosenfeld, A.; Levkin, P.A. Marrying chemistry with biology by combining on-chip solution-based combinatorial synthesis and cellular screening. Nat. Commun. 2019, 10, 2879. [Google Scholar] [CrossRef]
- Wong, Y.H.; Chiu, C.C.; Lin, C.L.; Chen, T.S.; Jheng, B.R.; Lee, Y.C.; Chen, J.; Chen, B.S. A New Era for Cancer Target Therapies: Applying Systems Biology and Computer-Aided Drug Design to Cancer Therapies. Curr. Pharm. Biotechnol. 2016, 17, 1246–1267. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Known Target | Inhibitor Class | Indications |
|---|---|---|---|
| Anaplastic lymphoma kinase (ALK) | |||
| Alectinib | ALK and RET | II | ALK+ NSCLC |
| Brigatinib | ALK, ROS1, IGF-1R, Flt3, EGFR | I | ALK+ NSCLC after crizotinib |
| Ceritinib | ALK, IGF-1R, InsR, ROS1 | II | ALK+ NSCLC as first-line treatment or after crizotinib resistance |
| Crizotinib | ALK, c-Met (HGFR), ROS1, MST1R | II | ALK+, ROS1+ NSCLC |
| Entrectinib | TRKA/B/C, ROS1, ALK | I | ROS1+ NSCLC; solid tumors with NTRK fusion proteins |
| Lorlatinib | ALK | I | ALK+ NSCLC |
| Fusion of breakpoint cluster region and Abelson (BCR-ABL) | |||
| Bosutinib | BCR-ABL, Src, Lyn, Hck | CML | |
| Dasatinib | BCR-ABL, EGFR, Src, Lck, Yes, Fyn, Kit, EphA2, PDGFRβ | I | Ph+ chronic ML and ALL |
| Imatinib | BCR-ABL, Kit, PDGFR | II | Ph+ CML or ALL, CEL, DFSP, HES, GIST, MDS/MDP |
| Nilotinib | BCR-ABL, PDGFR, DDR1 | II | Ph+ CLL |
| Ponatinib | BCR-ABL, BCR-ABL T315I, VEGFR, PDGFR, FGFR, EphR, Src family kinases, Kit, RET, Tie2, Flt3 | II | Ph+ CML or ALL |
| Epidermal growth factor receptor (EGFR) | |||
| Afatinib | EGFR, ErbB2, ErbB4 | Covalent (V) | NSCLC |
| Dacomitinib | EGFR/ErbB2/ErbB4 | I | EGFR- mutated NSCLC |
| Erlotinib | EGFR | I | SCLC and PaC |
| Gefitinib | EGFR | I | NCLC |
| Lapatinib | EGFR, ErbB2 | II | BC |
| Neratinib | ErbB2/HER2 | Covalent (V) | HER2+ breast cancer |
| Osimertinib | EGFR T970M | Covalent (V) | NSCLC |
| Vandetanib | EGFRs, VEGFRs, RET, Brk, Tie2, EphRs, Src family kinases | I | MTC |
| FMS-like tyrosine kinase 3 (FLT3) | |||
| Gilteritinib | FLT3 | I | AMLwith FLT3 mutation5 |
| Midostaurin | FLT3 | I | ALL Flt3 mutation+ |
| Fibroblast growth factor receptors (FGFR) | |||
| Erdafitinib | FGFR1/2/3/4 | I | Urothelial carcinoma |
| Janus kinase (JAK) | |||
| Ruxolitinib | JAK1 and 2 | I | MF and PV |
| Neurotrophic Tyrosine Receptor Kinase (NTRK) | |||
| Larotrectinib | NTRK | I | Solid tumors with NTRK gene fusion proteins |
| Vascular endothelial growth factor (VEGFR) | |||
| Axitinib | VEGFR1/2/3, PDGFRβ | II | RCC |
| Carbozantinib | RET, Met, VEGFR1/2/3, Kit, TrkB, Flt3, Axl, Tie2, ROS1 | I | Metastatic MTC, advanced RCC and HCC |
| Lenvatinib | VEGFRs, FGFRs, PDGFR, Kit, RET | II | DTC |
| Pazopanib | VEGFR1/2/3, PDGFRα/β, FGFR1/3, Kit, Lck, Fms, Itk | I | RCC, STS |
| Regorafenib | VEGFR1/2/3, BCR-ABL, BRAF, BRAF(V600E), Kit, PDGFRα/β, RET, FGFR1/2, Tie2, Eph2A | II | CRC, GIST |
| Sorafenib | B/C-Raf, BRAF (V600E), Kit, Flt3, RET, VEGFR1/2/3, PDGFRβ | II | RCC, DTC and HCC |
| Sunitinib | PDGFRα/β, VEGFR1/2/3, Kit, Flt3, CSF-1R, RET | II | RCC, GIST, PNET |
| BRAF | |||
| Dabrafenib | BRAF | I | Melanoma and NSCLC with BRAF mutations |
| Encorafenib | BRAFV600E/K | I | BRAFV600E/K mutant melanoma with binimetinib |
| Vemurafenib | A/B/C-Raf, BRAF (V600E), SRMS, ACK1, MAP4K5, FGR | I | Melanoma with BRAFV600E mutation and ECD |
| Bruton tyrosine kinase | |||
| Acalabrutinib | Bruton tyrosine kinase | Covalent (V) | MCL |
| Ibrutinib | Bruton tyrosine kinase | Covalent (V) | MCL, CLL, WM, graph vs host disease. |
| Mitogen-activated protein kinase kinase (MEK) | |||
| Binimetinib | MEK1/2 | III | BRAF V600E/K melanoma with encorafenib |
| Cobimetinib | MEK1/2 | III | Melanoma with BRAF V600E/K mutations with vemurafenib |
| Trametinib | MEK1/2 | III | Melanoma (2013) and NSCLC (2017) with BRAF mutations |
| Cyclin-dependent-kinase 4/6 | |||
| Abemaciclib | CDK4/6 | I | HR+, HER– BC |
| Palbociclib | CDK4/6 | I | ER+ and HER2– BC |
| Ribociclib | CDK4/6 | I | HR+-EGFR– metastatic BC |
| RTKI actions in favor of an anti-tumor immune response | ||
| RTKIs | Effects on immune cells | Characteristics of carried-out studies |
| BRAF inhibitors +/- MEK inhibitors | ↑ CD8+ TIL and melanoma antigen expression | Patient biopsies and in vivo pre-clinical study, BRAF mutated melanoma |
| Cabozantinib | ↑ neutrophil-mediated antitumor innate immunity | In vivo pre-clinical study, murine prostate cancer |
| Dasatinib | ↓ MDSCs | Patient biopsies and in vivo pre-clinical study, CML |
| Sorafenib | ↓ MDSCs | Patient biopsies and in vivo pre-clinical study, HCC |
| FGFR inhibitors | ↓ MDSCs | In vivo pre-clinical study, murine breast cancer |
| Sunitinib | ↓ MDSCs and M2 macrophages | In vivo pre-clinical study, RCC |
| VEGFR1 inhibitors | ↓ MDSCs, Tregs and M2 macrophages | In vivo pre-clinical studies on RCC and NSCLC |
| Tumor immune tolerance observed during the acquisition of resistance to RTKI | ||
| RTKI | Effects on immune cells | Characteristics of studies carried out |
| Axitinib | ↑ Tregs, ↑ and PD-1 expression | In vivo pre-clinical study, glioblastoma |
| BRAF inhibitors | ↑ MDSCs | In vivo pre-clinical study, BRAF mutated melanoma |
| Imatinib | ↑ M2 macrophages | In vivo pre-clinical study, GIST |
| Combinations of RTKI and checkpoint inhibitors under investigation | ||
| RTKI | Checkpoint inhibitors | Clinical trial |
| Dabrafenib | Pembrolizumab (anti-PD1) | Phase-2 trial, B-ref mutated melanoma |
| Lenvatinib | Pembrolizumab (anti-PD1) | Phase-2 trial, endometrial cancer and RCC |
| Regorafenib | Nivolumab | Phase-2 trial, gastric or colorectal cancer |
| Sunitinib | Atezolizumab (anti-PD-L1) | Phase-3 trial, metastatic RCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. https://doi.org/10.3390/cancers12030731
Pottier C, Fresnais M, Gilon M, Jérusalem G, Longuespée R, Sounni NE. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers. 2020; 12(3):731. https://doi.org/10.3390/cancers12030731
Chicago/Turabian StylePottier, Charles, Margaux Fresnais, Marie Gilon, Guy Jérusalem, Rémi Longuespée, and Nor Eddine Sounni. 2020. "Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy" Cancers 12, no. 3: 731. https://doi.org/10.3390/cancers12030731
APA StylePottier, C., Fresnais, M., Gilon, M., Jérusalem, G., Longuespée, R., & Sounni, N. E. (2020). Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers, 12(3), 731. https://doi.org/10.3390/cancers12030731