Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma

 , ,
, ,  ,
,  , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
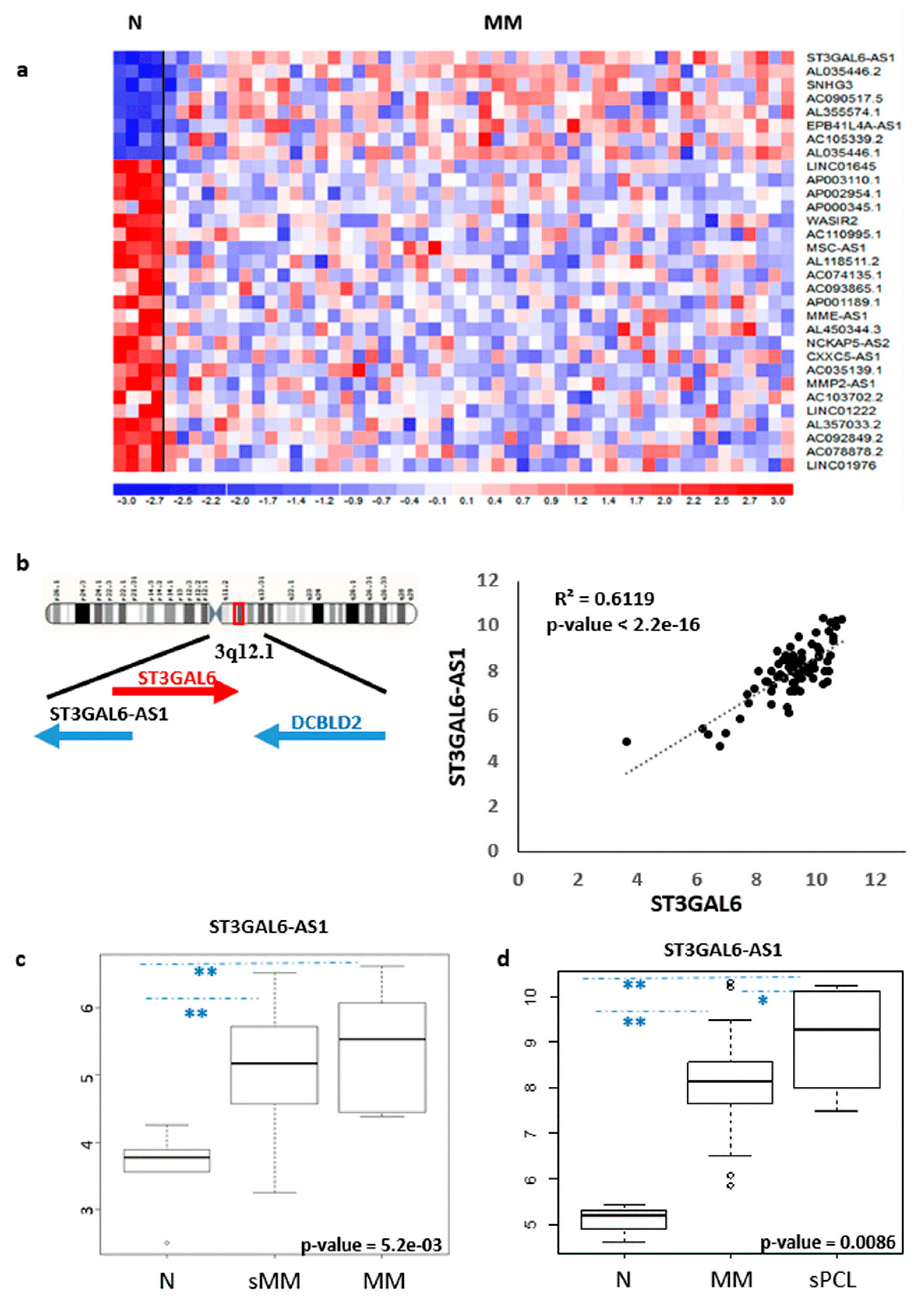
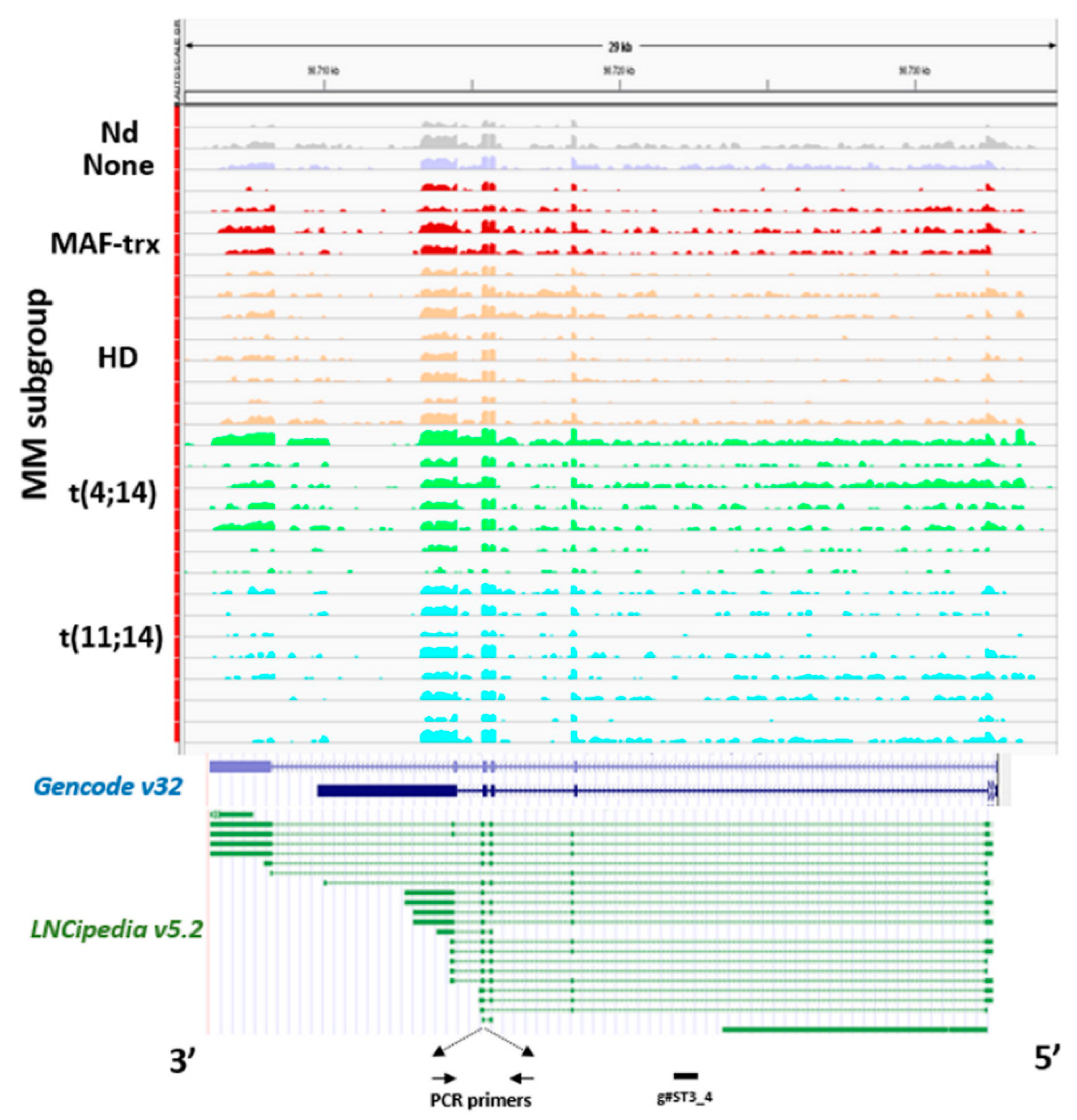
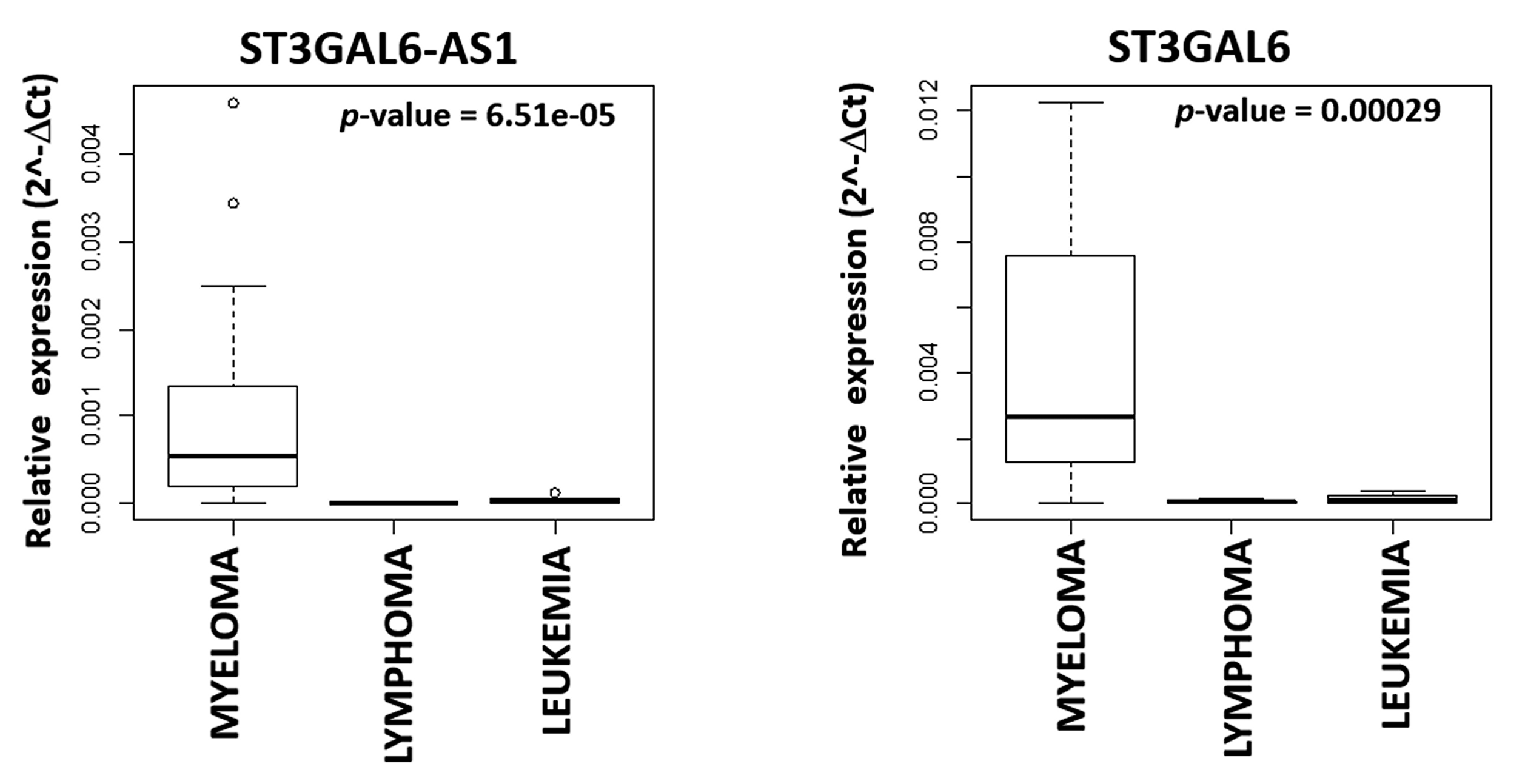
2.1. LncRNA ST3GAL6-AS1 is Overexpressed in MM Patients
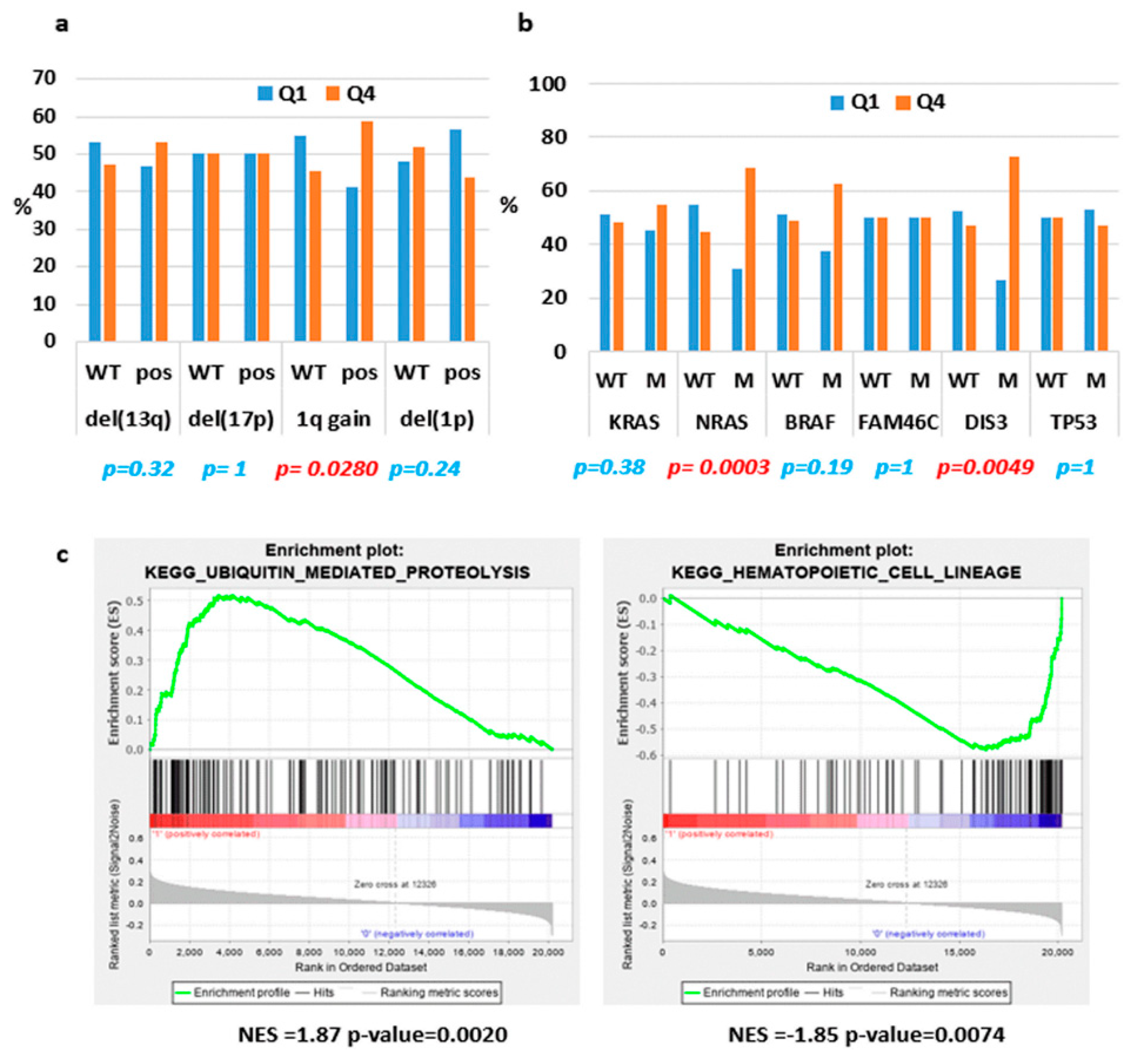
2.2. ST3GAL6-AS1 Expression is Unrelated to MM Prognosis
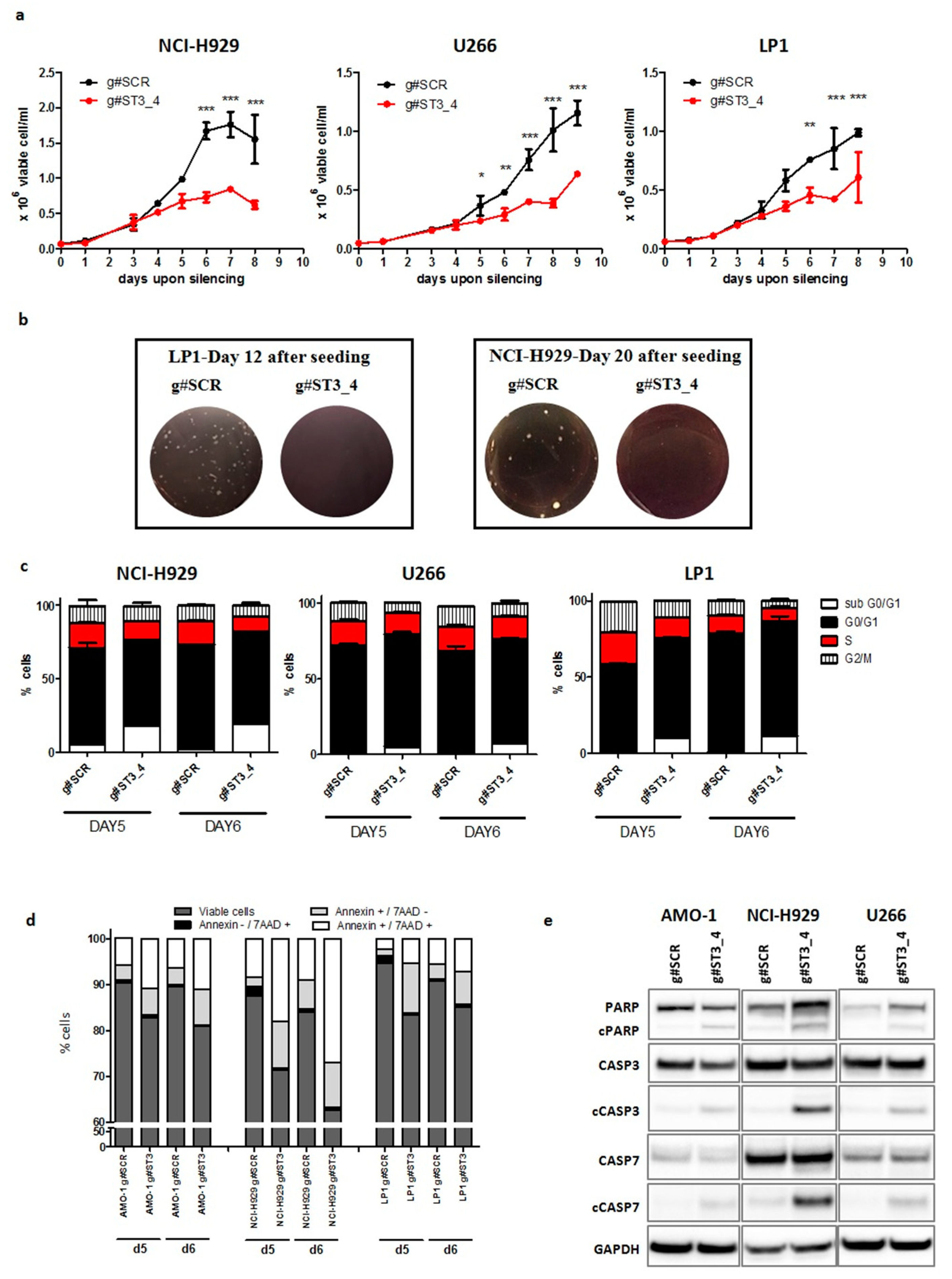
2.3. ST3GAL6-AS1 Silencing Affects Cell Cycle and Induces Apoptosis
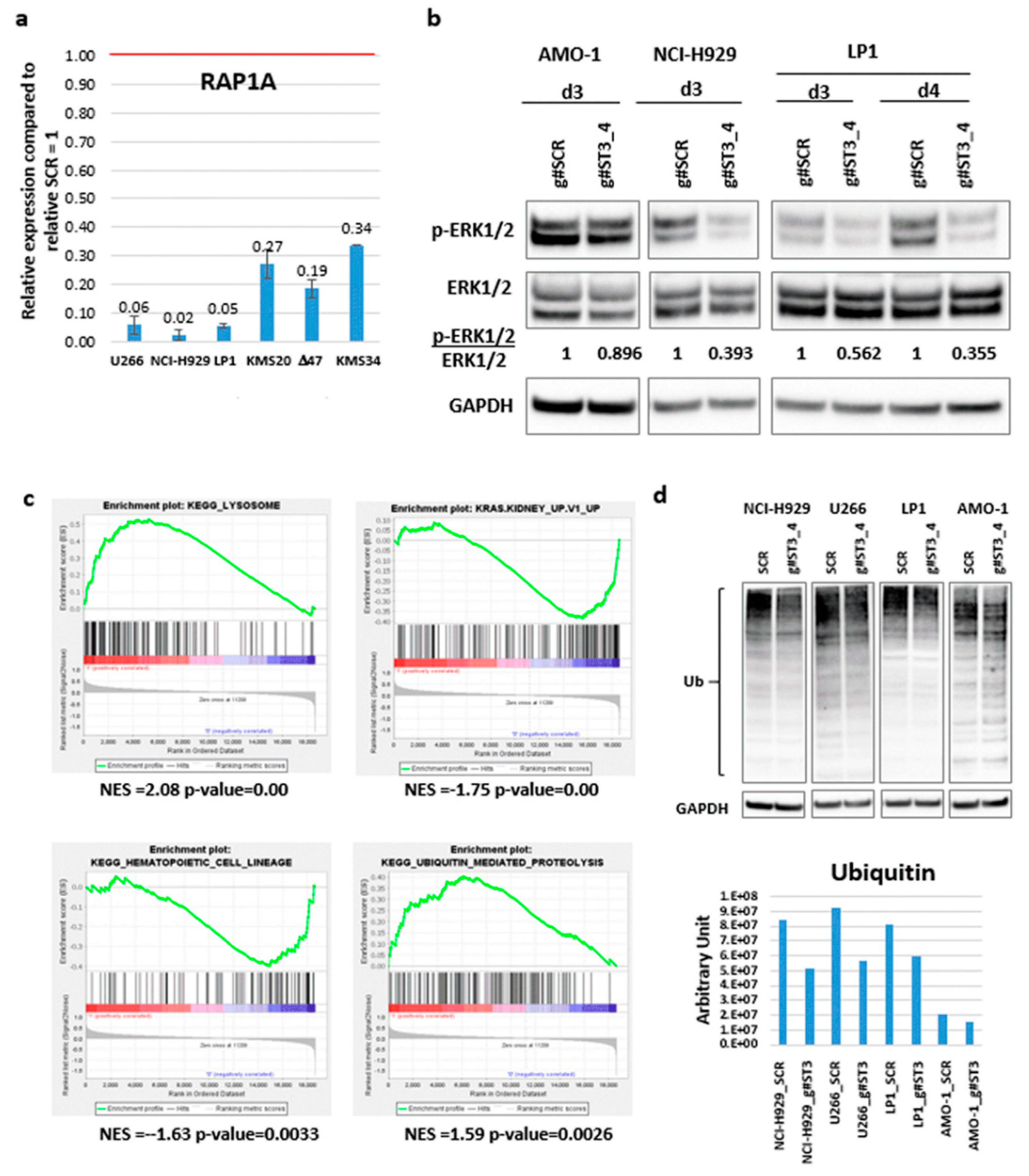
2.4. Identification of ST3GAL6-AS1 Targets in MM Cells
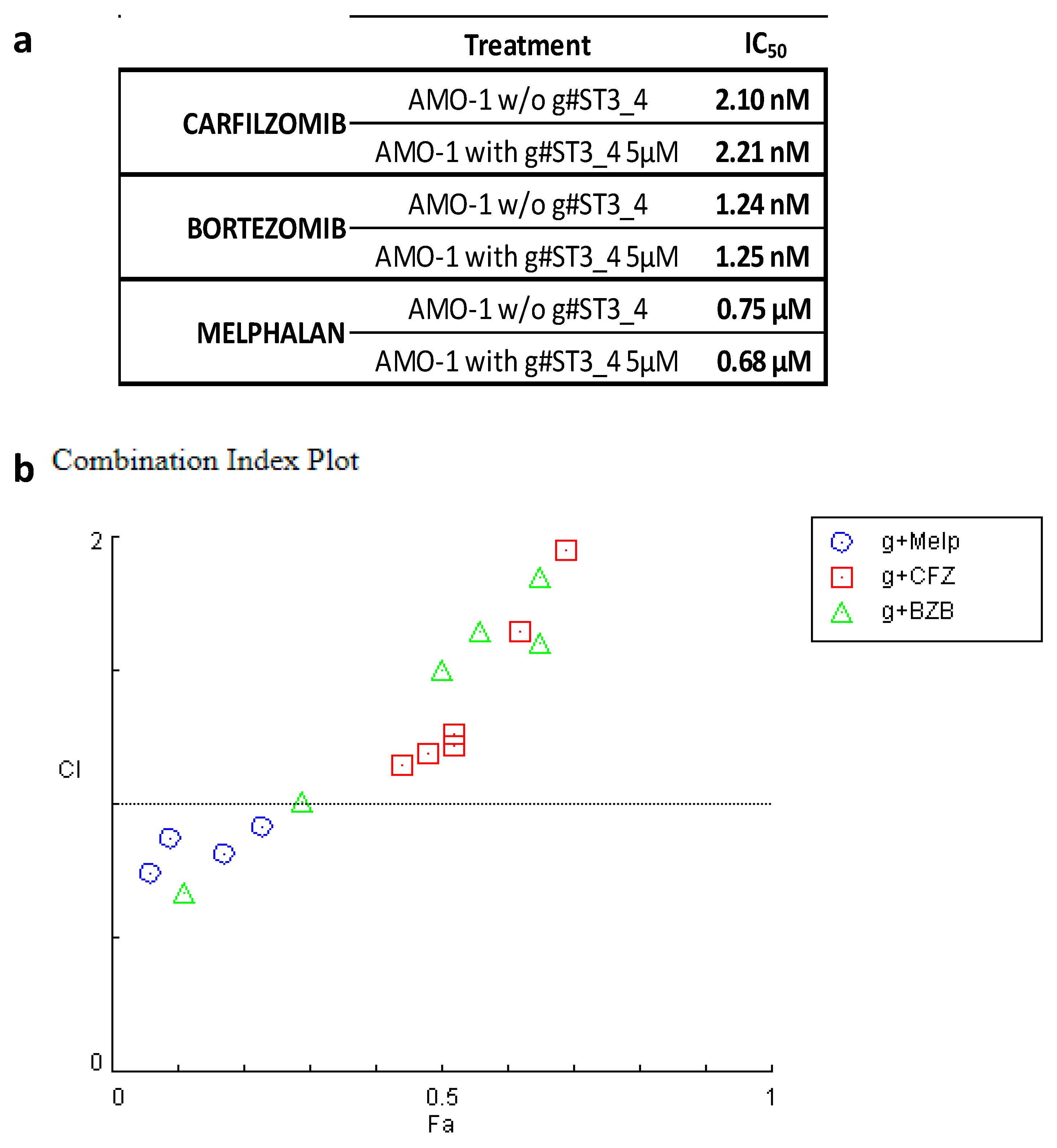
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Gymnosis
4.3. Colony Forming Assay
4.4. Cell Cycle Analysis and Apoptosis
4.5. Western Blot and Antibodies
4.6. Synergism Quantification
4.7. Gene Expression Profiling
4.8. RNA Sequencing (RNA-seq)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Morgan:, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Kunej, T.; Obsteter, J.; Pogacar, Z.; Horvat, S.; Calin, G.A. The decalog of long non-coding RNA involvement in cancer diagnosis and monitoring. Crit. Rev. Clin. Lab. Sci. 2014, 51, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butova, R.; Vychytilova-Faltejskova, P.; Souckova, A.; Sevcikova, S.; Hajek, R. Long Non-Coding RNAs in Multiple Myeloma. Non Coding RNA 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.S.; Song, Y.P.; Fang, B.J. The role of long non-coding RNAs in multiple myeloma. Eur. J. Haematol. 2019, 103, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Morelli, E.; Gulla, A.; Rocca, R.; Federico, C.; Raimondi, L.; Malvestiti, S.; Agosti, V.; Rossi, M.; Costa, G.; Giavaresi, G.; et al. The Non-Coding RNA Landscape of Plasma Cell Dyscrasias. Cancers 2020, 12, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, L.; Ronchetti, D.; Agnelli, L.; Taiana, E.; Vinci, C.; Neri, A. Long Non-Coding RNAs in Multiple Myeloma. Genes 2018, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Samur, M.K.; Minvielle, S.; Gulla, A.; Fulciniti, M.; Cleynen, A.; Aktas Samur, A.; Szalat, R.; Shammas, M.; Magrangeas, F.; Tai, Y.T.; et al. Long intergenic non-coding RNAs have an independent impact on survival in multiple myeloma. Leukemia 2018, 32, 2626–2635. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef] [Green Version]
- Handa, H.; Kuroda, Y.; Kimura, K.; Masuda, Y.; Hattori, H.; Alkebsi, L.; Matsumoto, M.; Kasamatsu, T.; Kobayashi, N.; Tahara, K.I.; et al. Long non-coding RNA MALAT1 is an inducible stress response gene associated with extramedullary spread and poor prognosis of multiple myeloma. Br. J. Haematol. 2017, 179, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Lin, J.; Fang, H.; Fang, J.; Li, C.; Chen, W.; Liu, S.; Ondrejka, S.; Gong, Z.; Reu, F.; et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018, 32, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Favasuli, V.; Ronchetti, D.; Todoerti, K.; Pelizzoni, F.; Manzoni, M.; Barbieri, M.; Fabris, S.; Silvestris, I.; Gallo Cantafio, M.E.; et al. Long non-coding RNA NEAT1 targeting impairs the DNA repair machinery and triggers anti-tumor activity in multiple myeloma. Leukemia 2020, 34, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Ronchetti, D.; Favasuli, V.; Todoerti, K.; Manzoni, M.; Amodio, N.; Tassone, P.; Agnelli, L.; Neri, A. Long non-coding RNA NEAT1 shows high expression unrelated to molecular features and clinical outcome in multiple myeloma. Haematologica 2019, 104, e72–e76. [Google Scholar] [CrossRef]
- Chen, L.; Hu, N.; Wang, C.; Zhao, H.; Gu, Y. Long non-coding RNA CCAT1 promotes multiple myeloma progression by acting as a molecular sponge of miR-181a-5p to modulate HOXA1 expression. Cell Cycle 2018, 17, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Zhang, Y.; Liu, W.; Huang, Y.; Shen, X.; Jing, R.; Pu, J.; Wang, X.; Ju, S.; Cong, H.; et al. LncRNA H19 overexpression induces bortezomib resistance in multiple myeloma by targeting MCL-1 via miR-29b-3p. Cell Death Dis. 2019, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Pan, J.; Zhang, N.; Wei, W.; Yu, S.; Ai, L. Knockdown of long non-coding RNA H19 inhibits multiple myeloma cell growth via NF-kappaB pathway. Sci. Rep. 2017, 7, 18079. [Google Scholar] [CrossRef] [Green Version]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Feng, Y.; Chen, H.; Huang, L.; Wang, F.; Bai, J.; Yang, Y.; Wang, J.; Zhao, W.; Jia, Y.; et al. Focusing on long non-coding RNA dysregulation in newly diagnosed multiple myeloma. Life Sci. 2018, 196, 133–142. [Google Scholar] [CrossRef]
- Hu, J.; Shan, Y.; Ma, J.; Pan, Y.; Zhou, H.; Jiang, L.; Jia, L. LncRNA ST3Gal6-AS1/ST3Gal6 axis mediates colorectal cancer progression by regulating alpha-2,3 sialylation via PI3K/Akt signaling. Int. J. Cancer 2019, 145, 450–460. [Google Scholar] [CrossRef]
- MMRF Researcher Gateway. Available online: https://research.themmrf.org (accessed on 10 October 2018).
- Ronchetti, D.; Agnelli, L.; Pietrelli, A.; Todoerti, K.; Manzoni, M.; Taiana, E.; Neri, A. A compendium of long non-coding RNAs transcriptional fingerprint in multiple myeloma. Sci. Rep. 2018, 8, 6557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deep Tools. Available online: https://deeptools.readthedocs.io/en/develop/index.html (accessed on 11 February 2018).
- IGV. Available online: http://www.broadinstitute.org/igv/ (accessed on 20 November 2019).
- Ronchetti, D.; Agnelli, L.; Taiana, E.; Galletti, S.; Manzoni, M.; Todoerti, K.; Musto, P.; Strozzi, F.; Neri, A. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget 2016, 7, 14814–14830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, A.; Ehrhardt, G.R.; Guo, X.; Schrader, J.W. Ras and relatives--job sharing and networking keep an old family together. Exp. Hematol. 2002, 30, 1089–1106. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, S.; Ronchetti, D.; Agnelli, L.; Baldini, L.; Morabito, F.; Bicciato, S.; Basso, D.; Todoerti, K.; Lombardi, L.; Lambertenghi-Deliliers, G.; et al. Transcriptional features of multiple myeloma patients with chromosome 1q gain. Leukemia 2007, 21, 1113–1116. [Google Scholar] [CrossRef]
- Agnelli, L.; Fabris, S.; Bicciato, S.; Basso, D.; Baldini, L.; Morabito, F.; Verdelli, D.; Todoerti, K.; Lambertenghi-Deliliers, G.; Lombardi, L.; et al. Upregulation of translational machinery and distinct genetic subgroups characterise hyperdiploidy in multiple myeloma. Br. J. Haematol. 2007, 136, 565–573. [Google Scholar] [CrossRef]
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Baldini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An Italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353–21361. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Fabris, S.; Tonon, G.; Segalla, S.; Cifola, I.; Pinatel, E.; Tassone, P.; et al. A compendium of DIS3 mutations and associated transcriptional signatures in plasma cell dyscrasias. Oncotarget 2015, 6, 26129–26141. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Marzorati, S.; Fabris, S.; Ciceri, G.; Galletti, S.; Milesi, G.; Manzoni, M.; et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: Implication for MEK-ERK pathway activation. Oncotarget 2015, 6, 24205–24217. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- GEO. Proprietary Dataset. Available online: http//www.ncbi.nlm.nih.gov/geo (accessed on 20 November 2019).







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronchetti, D.; Todoerti, K.; Vinci, C.; Favasuli, V.; Agnelli, L.; Manzoni, M.; Pelizzoni, F.; Chiaramonte, R.; Platonova, N.; Giuliani, N.; et al. Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma. Cancers 2020, 12, 782. https://doi.org/10.3390/cancers12040782
Ronchetti D, Todoerti K, Vinci C, Favasuli V, Agnelli L, Manzoni M, Pelizzoni F, Chiaramonte R, Platonova N, Giuliani N, et al. Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma. Cancers. 2020; 12(4):782. https://doi.org/10.3390/cancers12040782
Chicago/Turabian StyleRonchetti, Domenica, Katia Todoerti, Cristina Vinci, Vanessa Favasuli, Luca Agnelli, Martina Manzoni, Francesca Pelizzoni, Raffaella Chiaramonte, Natalia Platonova, Nicola Giuliani, and et al. 2020. "Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma" Cancers 12, no. 4: 782. https://doi.org/10.3390/cancers12040782
APA StyleRonchetti, D., Todoerti, K., Vinci, C., Favasuli, V., Agnelli, L., Manzoni, M., Pelizzoni, F., Chiaramonte, R., Platonova, N., Giuliani, N., Tassone, P., Amodio, N., Neri, A., & Taiana, E. (2020). Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma. Cancers, 12(4), 782. https://doi.org/10.3390/cancers12040782