Morphologic and Genomic Heterogeneity in the Evolution and Progression of Breast Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genomic Diversity of Primary Breast Cancer
3. Subclonal Genomic Diversity in Primary Breast Cancer
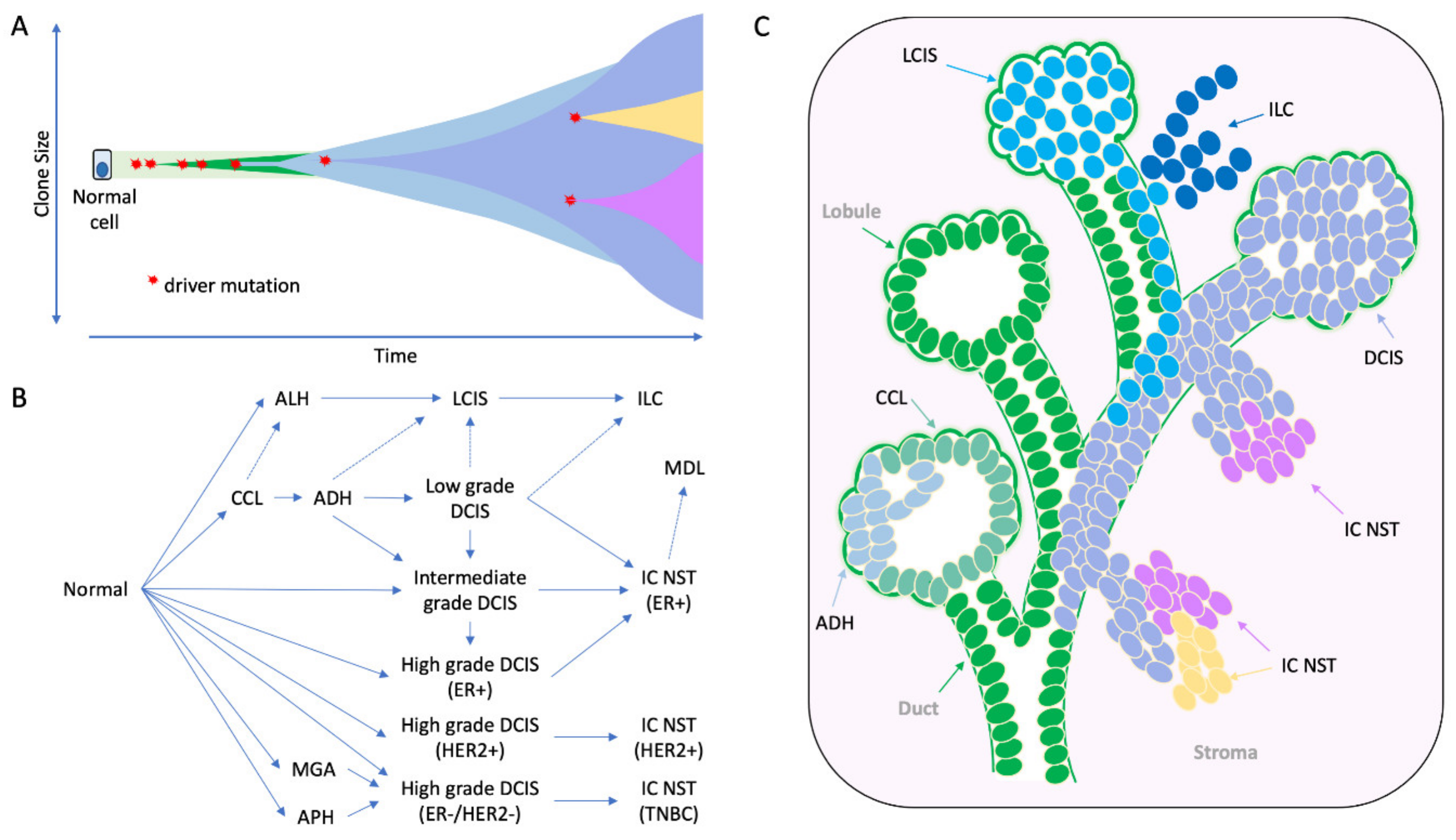
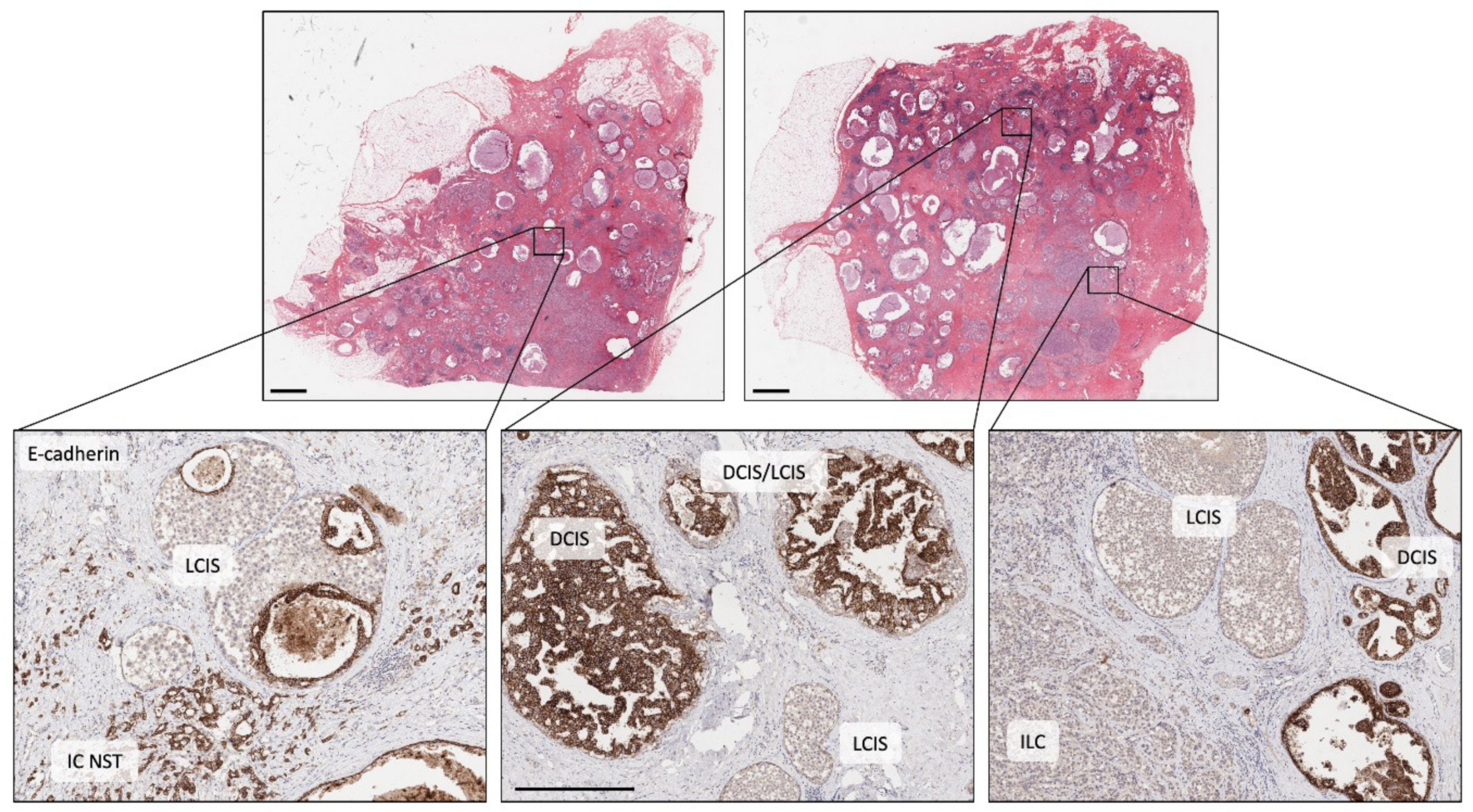
4. The Early Clonal Nature of Breast Cancer—Going Back to the Beginning
5. Genomics and Clonal Dynamic Changes During Metastatic Progression
6. Capturing Intra-Tumour Heterogeneity in Tissue or Liquid Biopsy
7. Clinical Implications and Utility in Breast Cancer
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. World Health Organisation Classification of Tumours: Breast Tumours, 5th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2019. [Google Scholar]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, J.C.; Nadeem, S.; Levine, A.J.; Pouryahya, M.; Deasy, J.O.; Tannenbaum, A. Robust and interpretable PAM50 reclassification exhibits survival advantage for myoepithelial and immune phenotypes. NPJ Breast Cancer 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Davies, H.; Morganella, S.; Purdie, C.A.; Jang, S.J.; Borgen, E.; Russnes, H.; Glodzik, D.; Zou, X.; Viari, A.; Richardson, A.L.; et al. Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017, 77, 4755–4762. [Google Scholar] [CrossRef] [Green Version]
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Vincent-Salomon, A.; Pivot, X.; Sertier, A.S.; Thomas, E.; Tonon, L.; Boyault, S.; Mulugeta, E.; Treilleux, I.; MacGrogan, G.; et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat. Commun. 2016, 7, 12222. [Google Scholar] [CrossRef]
- Hicks, J.; Krasnitz, A.; Lakshmi, B.; Navin, N.E.; Riggs, M.; Leibu, E.; Esposito, D.; Alexander, J.; Troge, J.; Grubor, V.; et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006, 16, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The contribution of germline predisposition gene mutations to clinical subtypes of invasive breast cancer from a clinical genetic testing cohort. J. Natl. Cancer Inst. 2020, djaa023. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Bi, R.; Kumar, R.; Blecua, P.; Mandelker, D.L.; Geyer, F.C.; Pareja, F.; James, P.A.; kConFab, I.; Couch, F.J.; et al. The Landscape of Somatic Genetic Alterations in Breast Cancers from ATM Germline Mutation Carriers. J. Natl. Cancer Inst. 2018, 110, 1030–1034. [Google Scholar] [CrossRef] [Green Version]
- Mandelker, D.; Kumar, R.; Pei, X.; Selenica, P.; Setton, J.; Arunachalam, S.; Ceyhan-Birsoy, O.; Brown, D.N.; Norton, L.; Robson, M.E.; et al. The Landscape of Somatic Genetic Alterations in Breast Cancers from CHEK2 Germline Mutation Carriers. JNCI Cancer Spectr. 2019, 3, pkz027. [Google Scholar] [CrossRef]
- Wilson, J.R.; Bateman, A.C.; Hanson, H.; An, Q.; Evans, G.; Rahman, N.; Jones, J.L.; Eccles, D.M. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J. Med. Genet. 2010, 47, 771–774. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Intra, M.; Trentin, C.; Veronesi, P.; Galimberti, V. CDH1 germline mutations and hereditary lobular breast cancer. Fam. Cancer 2016, 15, 215–219. [Google Scholar] [CrossRef]
- Fusco, N.; Geyer, F.C.; De Filippo, M.R.; Martelotto, L.G.; Ng, C.K.; Piscuoglio, S.; Guerini-Rocco, E.; Schultheis, A.M.; Fuhrmann, L.; Wang, L.; et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod. Pathol. 2016, 29, 1292–1305. [Google Scholar] [CrossRef]
- Miyai, K.; Schwartz, M.R.; Divatia, M.K.; Anton, R.C.; Park, Y.W.; Ayala, A.G.; Ro, J.Y. Adenoid cystic carcinoma of breast: Recent advances. World J. Clin. Cases 2014, 2, 732–741. [Google Scholar] [CrossRef]
- Letessier, A.; Ginestier, C.; Charafe-Jauffret, E.; Cervera, N.; Adelaide, J.; Gelsi-Boyer, V.; Ahomadegbe, J.C.; Benard, J.; Jacquemier, J.; Birnbaum, D.; et al. ETV6 gene rearrangements in invasive breast carcinoma. Genes Chromosomes Cancer 2005, 44, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Geyer, F.C.; Pareja, F.; Weigelt, B.; Rakha, E.; Ellis, I.O.; Schnitt, S.J.; Reis-Filho, J.S. The Spectrum of Triple-Negative Breast Disease: High- and Low-Grade Lesions. Am. J. Pathol. 2017, 187, 2139–2151. [Google Scholar] [PubMed] [Green Version]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothe, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCart Reed, A.E.; Kalaw, E.; Nones, K.; Bettington, M.; Lim, M.; Bennett, J.; Johnstone, K.; Kutasovic, J.R.; Saunus, J.M.; Kazakoff, S.; et al. Phenotypic and molecular dissection of Metaplastic Breast Cancer and the prognostic implications. J. Pathol. 2019, 247, 214–227. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Piscuoglio, S.; Geyer, F.C.; Burke, K.A.; Pareja, F.; Eberle, C.A.; Lim, R.S.; Natrajan, R.; Riaz, N.; Mariani, O.; et al. The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin. Cancer Res. 2017, 23, 3859–3870. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. NPJ Breast Cancer 2017, 3, 48. [Google Scholar]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; McNamara, K.; Reiter, J.G.; Sun, R.; Hu, Z.; Ma, Z.; Ding, J.; Suarez, C.J.; Tilk, S.; Raghavendra, A.; et al. Clonal replacement and heterogeneity in breast tumors treated with neoadjuvant HER2-targeted therapy. Nat. Commun. 2019, 10, 657. [Google Scholar] [CrossRef]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Kutasovic, J.R.; Nones, K.; Saunus, J.M.; Da Silva, L.; Newell, F.; Kazakoff, S.; Melville, L.; Jayanthan, J.; Vargas, A.C.; et al. Mixed ductal-lobular carcinomas: Evidence for progression from ductal to lobular morphology. J. Pathol. 2018, 244, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Fumagalli, D.; Pietri, E.; Zoppoli, G.; Brown, D.; Nik-Zainal, S.; Gundem, G.; Rothe, F.; Majjaj, S.; Garuti, A.; et al. Uncovering the genomic heterogeneity of multifocal breast cancer. J. Pathol. 2015, 236, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, P.T.; Gale, T.; Reis-Filho, J.S.; Jones, C.; Parry, S.; Sloane, J.P.; Hanby, A.; Pinder, S.E.; Lee, A.H.; Humphreys, S.; et al. Columnar cell lesions of the breast: The missing link in breast cancer progression? A morphological and molecular analysis. Am. J. Surg. Pathol. 2005, 29, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Troxell, M.L.; Brunner, A.L.; Neff, T.; Warrick, A.; Beadling, C.; Montgomery, K.; Zhu, S.; Corless, C.L.; West, R.B. Phosphatidylinositol-3-kinase pathway mutations are common in breast columnar cell lesions. Mod. Pathol. 2012, 25, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabbs, D.J.; Carter, G.; Fudge, M.; Peng, Y.; Swalsky, P.; Finkelstein, S. Molecular alterations in columnar cell lesions of the breast. Mod. Pathol. 2006, 19, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kader, T.; Hill, P.; Zethoven, M.; Goode, D.L.; Elder, K.; Thio, N.; Doyle, M.; Semple, T.; Sufyan, W.; Byrne, D.J.; et al. Atypical ductal hyperplasia is a multipotent precursor of breast carcinoma. J. Pathol. 2019, 248, 326–338. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.; Powe, D.G.; Hodi, Z.; Lee, A.H.; Reis-Filho, J.S.; Ellis, I.O. High frequency of coexistence of columnar cell lesions, lobular neoplasia, and low grade ductal carcinoma in situ with invasive tubular carcinoma and invasive lobular carcinoma. Am. J. Surg. Pathol. 2007, 31, 417–426. [Google Scholar] [CrossRef]
- Simpson, P.T.; Reis-Filho, J.S.; Gale, T.; Lakhani, S.R. Molecular evolution of breast cancer. J. Pathol. 2005, 205, 248–254. [Google Scholar] [CrossRef]
- Lopez-Garcia, M.A.; Geyer, F.C.; Lacroix-Triki, M.; Marchio, C.; Reis-Filho, J.S. Breast cancer precursors revisited: Molecular features and progression pathways. Histopathology 2010, 57, 171–192. [Google Scholar] [CrossRef]
- Bombonati, A.; Sgroi, D.C. The molecular pathology of breast cancer progression. J. Pathol. 2011, 223, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Allred, D.C.; Wu, Y.; Mao, S.; Nagtegaal, I.D.; Lee, S.; Perou, C.M.; Mohsin, S.K.; O’Connell, P.; Tsimelzon, A.; Medina, D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin. Cancer Res. 2008, 14, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Schizas, M.; Geyer, F.C.; Selenica, P.; Piscuoglio, S.; Sakr, R.A.; Ng, C.K.Y.; Carniello, J.V.S.; Towers, R.; Giri, D.D.; et al. Lobular Carcinomas in Situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin. Cancer Res. 2019, 25, 674–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelotto, L.G.; Baslan, T.; Kendall, J.; Geyer, F.C.; Burke, K.A.; Spraggon, L.; Piscuoglio, S.; Chadalavada, K.; Nanjangud, G.; Ng, C.K.; et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat. Med. 2017, 23, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Weng, Z.; Spies, N.; Zhu, S.X.; Newburger, D.E.; Kashef-Haghighi, D.; Batzoglou, S.; Sidow, A.; West, R.B. Cell-lineage heterogeneity and driver mutation recurrence in pre-invasive breast neoplasia. Genome Med. 2015, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.L.; Sandle, J.; Jones, A.A.; Sofronis, A.; Patani, N.R.; Lakhani, S.R. Mapping loss of heterozygosity in normal human breast cells from BRCA1/2 carriers. Br. J. Cancer 2006, 95, 515–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, G.; Lu, Y.; Zlotnikov, G.; Thor, A.D.; Smith, H.S. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 1996, 274, 2057–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellsworth, D.L.; Ellsworth, R.E.; Love, B.; Deyarmin, B.; Lubert, S.M.; Mittal, V.; Shriver, C.D. Genomic patterns of allelic imbalance in disease free tissue adjacent to primary breast carcinomas. Breast Cancer Res. Treat. 2004, 88, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Bisoffi, M.; Fordyce, C.A.; Haaland, C.M.; Hines, W.C.; Joste, N.E.; Griffith, J.K. Telomere DNA content and allelic imbalance demonstrate field cancerization in histologically normal tissue adjacent to breast tumors. Int. J. Cancer 2006, 119, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Larson, P.S.; Schlechter, B.L.; de las Morenas, A.; Garber, J.E.; Cupples, L.A.; Rosenberg, C.L. Allele imbalance, or loss of heterozygosity, in normal breast epithelium of sporadic breast cancer cases and BRCA1 gene mutation carriers is increased compared with reduction mammoplasty tissues. J. Clin. Oncol. 2005, 23, 8613–8619. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz-Yildirim, M.; Silberman, R.E.; Langenbucher, A.; Saladi, S.V.; Ross, K.N.; Zarcaro, E.; Desmond, A.; Yildirim, M.; Vivekanandan, V.; Ravichandran, H.; et al. Aneuploidy and a deregulated DNA damage response suggest haploinsufficiency in breast tissues of BRCA2 mutation carriers. Sci. Adv. 2020, 6, eaay2611. [Google Scholar] [CrossRef] [Green Version]
- Konishi, H.; Mohseni, M.; Tamaki, A.; Garay, J.P.; Croessmann, S.; Karnan, S.; Ota, A.; Wong, H.Y.; Konishi, Y.; Karakas, B.; et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17773–17778. [Google Scholar] [CrossRef] [Green Version]
- Sedic, M.; Skibinski, A.; Brown, N.; Gallardo, M.; Mulligan, P.; Martinez, P.; Keller, P.J.; Glover, E.; Richardson, A.L.; Cowan, J.; et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun. 2015, 6, 7505. [Google Scholar] [CrossRef]
- Tan, M.P.; Tot, T. The sick lobe hypothesis, field cancerisation and the new era of precision breast surgery. Gland Surg. 2018, 7, 611–618. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, M.B.; He, X.; Hoadley, K.A.; Hoyle, A.; Pearce, J.B.; Garrett, A.L.; Kumar, S.; Moylan, V.J.; Brady, C.M.; Van Swearingen, A.E.; et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Investig. 2018, 128, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mattos-Arruda, L.; Sammut, S.J.; Ross, E.M.; Bashford-Rogers, R.; Greenstein, E.; Markus, H.; Morganella, S.; Teng, Y.; Maruvka, Y.; Pereira, B.; et al. The Genomic and Immune Landscapes of Lethal Metastatic Breast Cancer. Cell Rep. 2019, 27, 2690–2708. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Siegel, M.B.; Kanchi, K.L.; Miller, C.A.; Ding, L.; Zhao, W.; He, X.; Parker, J.S.; Wendl, M.C.; Fulton, R.S.; et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016, 13, e1002174. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [Green Version]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.K.Y.; Bidard, F.C.; Piscuoglio, S.; Geyer, F.C.; Lim, R.S.; de Bruijn, I.; Shen, R.; Pareja, F.; Berman, S.H.; Wang, L.; et al. Genetic Heterogeneity in Therapy-Naive Synchronous Primary Breast Cancers and Their Metastases. Clin. Cancer Res. 2017, 23, 4402–4415. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutasovic, J.R.; McCart Reed, A.E.; Males, R.; Sim, S.; Saunus, J.M.; Dalley, A.; McEvoy, C.R.; Dedina, L.; Miller, G.; Peyton, S.; et al. Breast cancer metastasis to gynaecological organs: A clinico-pathological and molecular profiling study. J. Pathol. Clin. Res. 2019, 5, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, I.; Karthik, G.M.; Alkodsi, A.; Kjallquist, U.; Stalhammar, G.; Lovrot, J.; Martinez, N.F.; Lagergren, J.; Hautaniemi, S.; Hartman, J.; et al. Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Investig. 2018, 128, 1355–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurilio, G.; Disalvatore, D.; Pruneri, G.; Bagnardi, V.; Viale, G.; Curigliano, G.; Adamoli, L.; Munzone, E.; Sciandivasci, A.; De Vita, F.; et al. A meta-analysis of oestrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 discordance between primary breast cancer and metastases. Eur. J. Cancer 2014, 50, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Cummings, M.C.; Simpson, P.T.; Reid, L.E.; Jayanthan, J.; Skerman, J.; Song, S.; McCart Reed, A.E.; Kutasovic, J.R.; Morey, A.L.; Marquart, L.; et al. Metastatic progression of breast cancer: Insights from 50 years of autopsies. J. Pathol. 2014, 232, 23–31. [Google Scholar] [CrossRef] [Green Version]
- De Duenas, E.M.; Hernandez, A.L.; Zotano, A.G.; Carrion, R.M.; Lopez-Muniz, J.I.; Novoa, S.A.; Rodriguez, A.L.; Fidalgo, J.A.; Lozano, J.F.; Gasion, O.B.; et al. Prospective evaluation of the conversion rate in the receptor status between primary breast cancer and metastasis: Results from the GEICAM 2009-03 ConvertHER study. Breast Cancer Res. Treat. 2014, 143, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Niikura, N.; Liu, J.; Hayashi, N.; Mittendorf, E.A.; Gong, Y.; Palla, S.L.; Tokuda, Y.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Ueno, N.T. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J. Clin. Oncol. 2012, 30, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Lindstrom, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]
- Lluch, A.; Gonzalez-Angulo, A.M.; Casadevall, D.; Eterovic, A.K.; Martinez de Duenas, E.; Zheng, X.; Guerrero-Zotano, A.; Liu, S.; Perez, R.; Chen, K.; et al. Dynamic clonal remodelling in breast cancer metastases is associated with subtype conversion. Eur. J. Cancer 2019, 120, 54–64. [Google Scholar] [CrossRef]
- Saunus, J.M.; Quinn, M.C.; Patch, A.M.; Pearson, J.V.; Bailey, P.J.; Nones, K.; McCart Reed, A.E.; Miller, D.; Wilson, P.J.; Al-Ejeh, F.; et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J. Pathol. 2015, 237, 363–378. [Google Scholar] [CrossRef]
- Wu, J.M.; Fackler, M.J.; Halushka, M.K.; Molavi, D.W.; Taylor, M.E.; Teo, W.W.; Griffin, C.; Fetting, J.; Davidson, N.E.; De Marzo, A.M.; et al. Heterogeneity of breast cancer metastases: Comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin. Cancer Res. 2008, 14, 1938–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, K.M.; Priedigkeit, N.; Basudan, A.; Tasdemir, N.; Sikora, M.J.; Sokol, E.S.; Hartmaier, R.J.; Ding, K.; Ahmad, N.Z.; Watters, R.J.; et al. FGFR4 overexpression and hotspot mutations in metastatic ER+ breast cancer are enriched in the lobular subtype. NPJ Breast Cancer 2019, 5, 19. [Google Scholar] [CrossRef]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Meijer-van Gelder, M.E.; Sweep, F.C.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Smeets, D.; Szekely, B.; Larsimont, D.; Szasz, A.M.; Adnet, P.Y.; Rothe, F.; Rouas, G.; Nagy, Z.I.; Farago, Z.; et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 2017, 8, 14944. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, G.V.; Powell, E.; Seth, S.; Ge, Z.; Carugo, A.; Bristow, C.; Peoples, M.; Robinson, F.; Qiu, H.; Shao, J.; et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat. Commun. 2018, 9, 5079. [Google Scholar] [CrossRef] [Green Version]
- Savas, P.; Teo, Z.L.; Lefevre, C.; Flensburg, C.; Caramia, F.; Alsop, K.; Mansour, M.; Francis, P.A.; Thorne, H.A.; Silva, M.J.; et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLoS Med. 2016, 13, e1002204. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelletti, V.; Appierto, V.; Tiberio, P.; Fina, E.; Callari, M.; Daidone, M.G. Circulating Biomarkers for Prediction of Treatment Response. J. Natl. Cancer Inst. Monogr. 2015, 2015, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janni, W.J.; Rack, B.; Terstappen, L.W.; Pierga, J.Y.; Taran, F.A.; Fehm, T.; Hall, C.; de Groot, M.R.; Bidard, F.C.; Friedl, T.W.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583–2593. [Google Scholar] [CrossRef] [Green Version]
- Rack, B.; Schindlbeck, C.; Juckstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106, dju066. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Pierga, J.Y.; Reuben, J.; Rademaker, A.; Davis, A.A.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): International expert consensus paper. Crit. Rev. Oncol. Hematol. 2019, 134, 39–45. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, C.; Wan, S.; Mu, Z.; Zhang, Z.; Abu-Khalaf, M.M.; Fellin, F.M.; Silver, D.P.; Neupane, M.; Jaslow, R.J.; et al. Association of clinical outcomes in metastatic breast cancer patients with circulating tumour cell and circulating cell-free DNA. Eur. J. Cancer 2019, 106, 133–143. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Wallwiener, M.; Hartkopf, A.D.; Riethdorf, S.; Nees, J.; Sprick, M.R.; Schonfisch, B.; Taran, F.A.; Heil, J.; Sohn, C.; Pantel, K.; et al. The impact of HER2 phenotype of circulating tumor cells in metastatic breast cancer: A retrospective study in 107 patients. BMC Cancer 2015, 15, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehm, T.; Muller, V.; Aktas, B.; Janni, W.; Schneeweiss, A.; Stickeler, E.; Lattrich, C.; Lohberg, C.R.; Solomayer, E.; Rack, B.; et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: A prospective, multicenter trial. Breast Cancer Res. Treat. 2010, 124, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kruijff, I.E.; Sieuwerts, A.M.; Onstenk, W.; Jager, A.; Hamberg, P.; de Jongh, F.E.; Smid, M.; Kraan, J.; Timmermans, M.A.; Martens, J.W.M.; et al. Androgen receptor expression in circulating tumor cells of patients with metastatic breast cancer. Int. J. Cancer 2019, 145, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, A.; Topa, J.; Nagel, A.; Skokowski, J.; Seroczynska, B.; Stokowy, T.; Welnicka-Jaskiewicz, M.; Zaczek, A.J. Spectrum of Epithelial-Mesenchymal Transition Phenotypes in Circulating Tumour Cells from Early Breast Cancer Patients. Cancers 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Mu, Z.; Wang, C.; Ye, Z.; Austin, L.; Civan, J.; Hyslop, T.; Palazzo, J.P.; Jaslow, R.; Li, B.; Myers, R.E.; et al. Prospective assessment of the prognostic value of circulating tumor cells and their clusters in patients with advanced-stage breast cancer. Breast Cancer Res. Treat. 2015, 154, 563–571. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Appierto, V.; Di Cosimo, S.; Reduzzi, C.; Pala, V.; Cappelletti, V.; Daidone, M.G. How to study and overcome tumor heterogeneity with circulating biomarkers: The breast cancer case. Semin. Cancer Biol. 2017, 44, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [PubMed]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts. Clin. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.; Cristofanilli, M.; Fortina, P. Detection of Activating Estrogen Receptor Gene (ESR1) Mutations in Single Circulating Tumor Cells. Clin. Cancer Res. 2017, 23, 6086–6093. [Google Scholar] [CrossRef] [Green Version]
- Beije, N.; Sieuwerts, A.M.; Kraan, J.; Van, N.M.; Onstenk, W.; Vitale, S.R.; van der Vlugt-Daane, M.; Dirix, L.Y.; Brouwer, A.; Hamberg, P.; et al. Estrogen receptor mutations and splice variants determined in liquid biopsies from metastatic breast cancer patients. Mol. Oncol. 2018, 12, 48–57. [Google Scholar] [CrossRef]
- Desmedt, C.; Pingitore, J.; Rothe, F.; Marchio, C.; Clatot, F.; Rouas, G.; Richard, F.; Bertucci, F.; Mariani, O.; Galant, C.; et al. ESR1 mutations in metastatic lobular breast cancer patients. NPJ Breast Cancer 2019, 5, 9. [Google Scholar] [CrossRef]
- Chung, J.H.; Pavlick, D.; Hartmaier, R.; Schrock, A.B.; Young, L.; Forcier, B.; Ye, P.; Levin, M.K.; Goldberg, M.; Burris, H.; et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann. Oncol. 2017, 28, 2866–2873. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, Y.; Siddiqui, B.; Hu, J.; Pun, M.; Cornwell, M.; Buchwalter, G.; Hughes, M.E.; Wagle, N.; Kirschmeier, P.; Janne, P.A.; et al. Unraveling the clinicopathological features driving the emergence of ESR1 mutations in metastatic breast cancer. NPJ Breast Cancer 2018, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, F.; Bidard, F.C.; Houy, A.; Saliou, A.; Madic, J.; Rampanou, A.; Hego, C.; Milder, M.; Cottu, P.; Sablin, M.P.; et al. Patient-Specific Circulating Tumor DNA Detection during Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Clin. Chem. 2017, 63, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madic, J.; Kiialainen, A.; Bidard, F.C.; Birzele, F.; Ramey, G.; Leroy, Q.; Rio Frio, T.; Vaucher, I.; Raynal, V.; Bernard, V.; et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int. J. Cancer 2015, 136, 2158–2165. [Google Scholar] [CrossRef]
- Rothe, F.; Laes, J.F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Inao, T.; Sueta, A.; Fujiwara, S.; Omoto, Y.; Iwase, H. Clinical significance of monitoring ESR1 mutations in circulating cell-free DNA in estrogen receptor positive breast cancer patients. Oncotarget 2016, 7, 32504–32518. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Ma, F.; Zhu, W.; Guan, Y.; Yang, L.; Xia, X.; Chen, S.; Li, Q.; Guan, X.; Yi, Z.; Qian, H.; et al. ctDNA dynamics: A novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget 2016, 7, 66020–66031. [Google Scholar] [CrossRef]
- Rothe, F.; Silva, M.J.; Venet, D.; Campbell, C.; Bradburry, I.; Rouas, G.; de Azambuja, E.; Maetens, M.; Fumagalli, D.; Rodrik-Outmezguine, V.; et al. Circulating Tumor DNA in HER2-Amplified Breast Cancer: A Translational Research Substudy of the NeoALTTO Phase III Trial. Clin. Cancer Res. 2019, 25, 3581–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, A.; Dreau, H.; Knight, S.J.L.; Ridout, K.; Mizani, T.; Vavoulis, D.; Colling, R.; Antoniou, P.; Kvikstad, E.M.; Pentony, M.M.; et al. Clinically actionable mutation profiles in patients with cancer identified by whole-genome sequencing. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef] [Green Version]
- Pineiro-Yanez, E.; Reboiro-Jato, M.; Gomez-Lopez, G.; Perales-Paton, J.; Troule, K.; Rodriguez, J.M.; Tejero, H.; Shimamura, T.; Lopez-Casas, P.P.; Carretero, J.; et al. PanDrugs: A novel method to prioritize anticancer drug treatments according to individual genomic data. Genome Med. 2018, 10, 41. [Google Scholar] [CrossRef]
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy-leveraging a knowledge base for clinical decision-making. Cold Spring Harb. Mol. Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Johnson, A.; Holla, V.; Bailey, A.M.; Brusco, L.; Chen, K.; Routbort, M.; Patel, K.P.; Zeng, J.; Kopetz, S.; et al. A decision support framework for genomically informed investigational cancer therapy. J. Natl. Cancer Inst. 2015, djv098. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Yates, L.R.; Desmedt, C. Translational Genomics: Practical Applications of the Genomic Revolution in Breast Cancer. Clin. Cancer Res. 2017, 23, 2630–2639. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.Y.; Shen, Y.; Pleasance, E.; Kasaian, K.; Leelakumari, S.; Jones, M.; Bose, P.; Ch’ng, C.; Reisle, C.; Eirew, P.; et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 7521–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reutersward, C.; Hakkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.G.; Verkaik, N.S.; Sieuwerts, A.M.; van Riet, J.; Naipal, K.A.T.; van Deurzen, C.H.M.; den Bakker, M.A.; Sleddens, H.; Dubbink, H.J.; den Toom, T.D.; et al. Correction: Functional Ex Vivo Assay Reveals Homologous Recombination Deficiency in Breast Cancer Beyond BRCA Gene Defects. Clin. Cancer Res. 2019, 25, 2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Banerji, U.; Dean, E.J.; Perez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2018, 24, 2050–2059. [Google Scholar] [CrossRef] [Green Version]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutasovic, J.R.; McCart Reed, A.E.; Sokolova, A.; Lakhani, S.R.; Simpson, P.T. Morphologic and Genomic Heterogeneity in the Evolution and Progression of Breast Cancer. Cancers 2020, 12, 848. https://doi.org/10.3390/cancers12040848
Kutasovic JR, McCart Reed AE, Sokolova A, Lakhani SR, Simpson PT. Morphologic and Genomic Heterogeneity in the Evolution and Progression of Breast Cancer. Cancers. 2020; 12(4):848. https://doi.org/10.3390/cancers12040848
Chicago/Turabian StyleKutasovic, Jamie R., Amy E. McCart Reed, Anna Sokolova, Sunil R. Lakhani, and Peter T. Simpson. 2020. "Morphologic and Genomic Heterogeneity in the Evolution and Progression of Breast Cancer" Cancers 12, no. 4: 848. https://doi.org/10.3390/cancers12040848
APA StyleKutasovic, J. R., McCart Reed, A. E., Sokolova, A., Lakhani, S. R., & Simpson, P. T. (2020). Morphologic and Genomic Heterogeneity in the Evolution and Progression of Breast Cancer. Cancers, 12(4), 848. https://doi.org/10.3390/cancers12040848