Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma

 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Presentation of the Patients’ Cohort
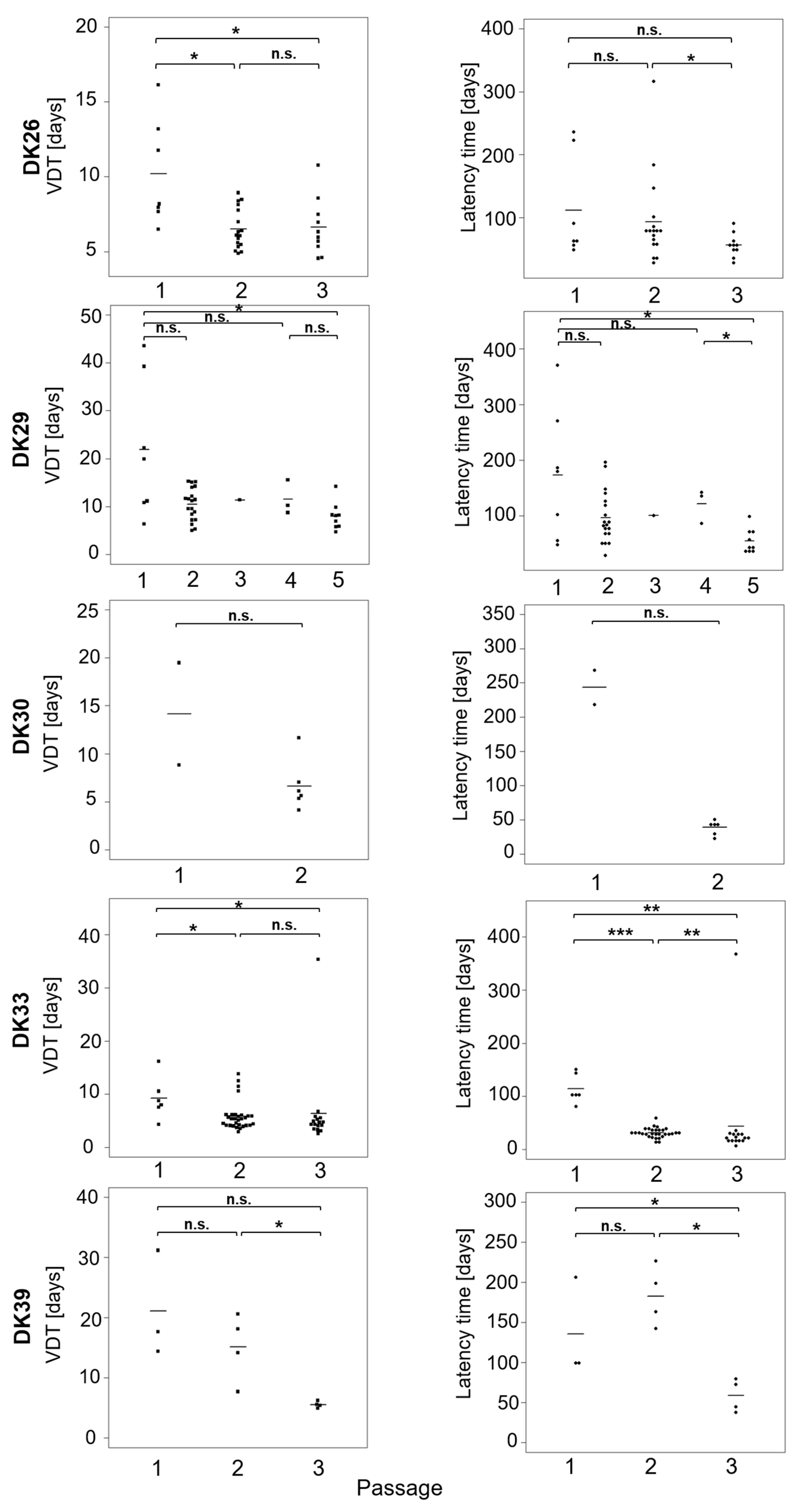
2.2. Take Rate in Nude Mouse and Tumour Measurements
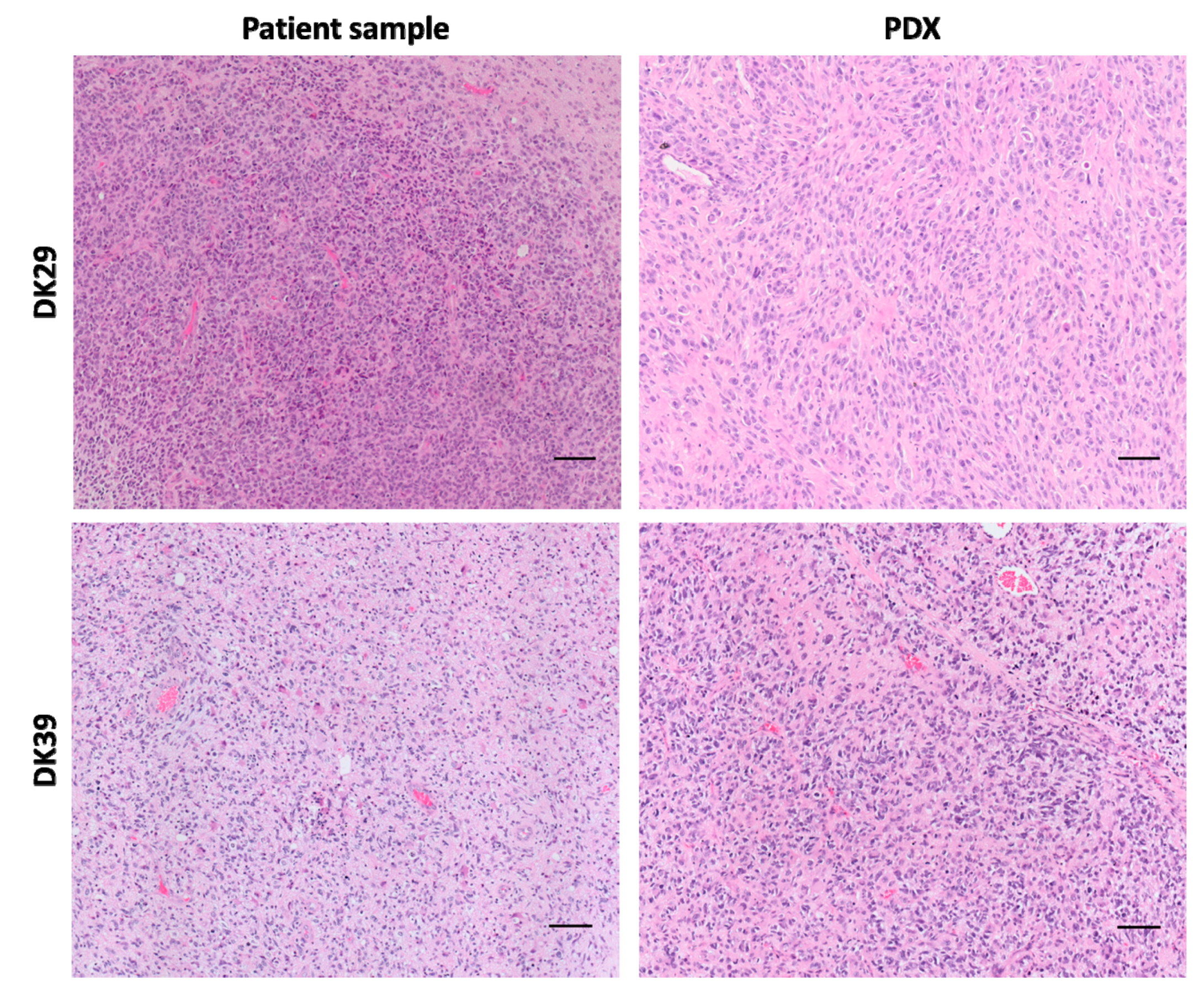
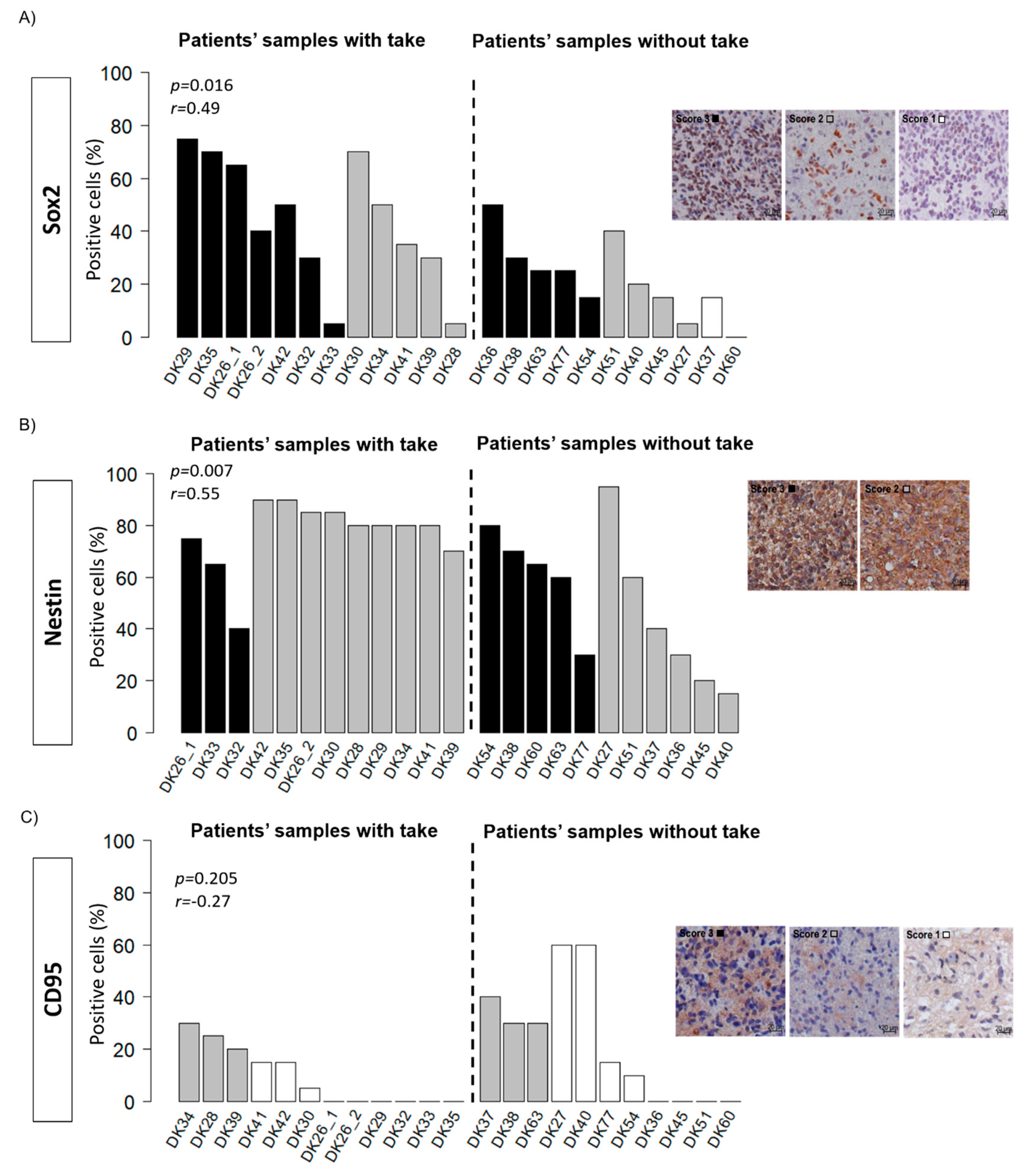
2.3. Histology and Putative Stem Cell Markers
2.4. Methylation of the MGMT Promoter
3. Discussion
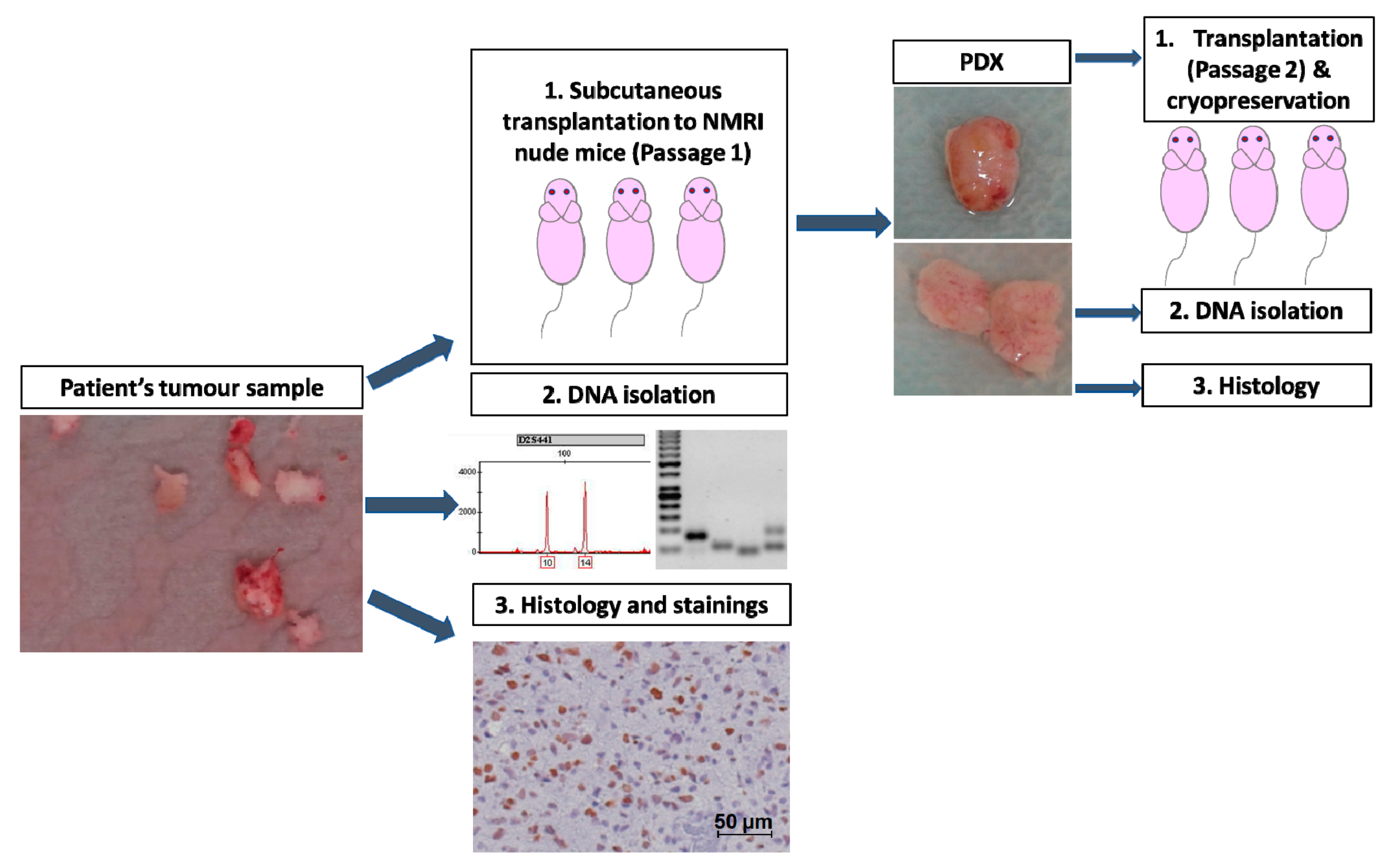
4. Materials and Methods
4.1. Human Tumour Samples
4.2. Animals
4.3. Stainings
4.3.1. Hematoxylin and Eosin
4.3.2. Putative Cancer Stem Cell Markers
4.4. DNA Isolation
4.5. Microsatellite Analysis
4.6. Methylation Specific PCR
4.7. Tumour Volume
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIC | Akaike Information Criterion |
| CD95 | Cluster of Differentiation 95 |
| CDX | Cell line derived xenograft |
| CSC | Cancer stem cell |
| DK | Deutsches Konsortium |
| MRI | Magnetic Resonance Imaging |
| MGMT | O6-MethylguaninDNA-Methyltransferase |
| MSP | Methylation-specific PCR |
| M | methylated template |
| U | unmethylated template |
| PDX | Patient-derived xenograft |
| RT | Radiotherapy |
| Sox2 | Sex determining region Y-box 2 |
| VDT | volume doubling time |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, O.; Ardanaz, E.; Botta, L.; Sant, M.; Tavilla, A.; Minicozzi, P. Survival of adults with primary malignant brain tumours in Europe; Results of the EUROCARE-5 study. Eur. J. Cancer 2015, 51, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in radiotherapy for glioblastoma. Front. Neurol. 2018, 8, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Dietrich, P.-Y.; Kraljevic, S.O.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef]
- Rivera, A.L.; Pelloski, C.E.; Gilbert, M.R.; Colman, H.; De La Cruz, C.; Sulman, E.P.; Bekele, B.N.; Aldape, K.D. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 2010, 12, 116–121. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; Vitis, S.D.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer stem cell hierarchy in glioblastoma multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Sathyan, P.; Zinn, P.O.; Marisetty, A.L.; Liu, B.; Kamal, M.M.; Singh, S.K.; Bady, P.; Lu, L.; Wani, K.M.; Veo, B.L.; et al. Mir-21–Sox2 Axis Delineates Glioblastoma Subtypes with Prognostic Impact. J. Neurosci. 2015, 35, 15097–15112. [Google Scholar] [CrossRef] [Green Version]
- Dahlstrand, J.; Collins, V.P.; Lendahl, U. Expression of the class VI intermediate filament nestin in human central nervous system tumors. Cancer Res. 1992, 52, 5334–5341. [Google Scholar]
- Drachsler, M.; Kleber, S.; Mateos, A.; Volk, K.; Mohr, N.; Chen, S.; Cirovic, B.; Tüttenberg, J.; Gieffers, C.; Sykora, J.; et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis. 2016, 7, e2209. [Google Scholar] [CrossRef] [PubMed]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taillandier, L.; Antunes, L.; Angioi-Duprez, K.S. Models for neuro-oncological preclinical studies: Solid orthotopic and heterotopic grafts of human gliomas into nude mice. J. Neurosci. Methods 2003, 125, 147–157. [Google Scholar] [CrossRef]
- Kanabur, P.; Guo, S.; Simonds, G.R.; Kelly, D.F.; Gourdie, R.G.; Verbridge, S.S.; Sheng, Z. Patient-derived glioblastoma stem cells respond differentially to targeted therapies. Patient-derived glioblastoma stem cells respond differentially to targeted therapies. Oncotarget 2016, 7, 86406–86419. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.-L.; Park, W.-Y.; Kong, D.-S.; et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; DuBois, W.; Pu, A.; Freeman, J.; Suit, H.D. Response of xenografts of human malignant gliomas and squamous cell carcinomas to fractionated irradiation. Int. J. Radiat. Oncol. 1992, 23, 803–809. [Google Scholar] [CrossRef]
- Taghian, A.; Dubois, W.; Budach, W.; Baumann, M.; Freeman, J.; Suit, H. In vivo radiation sensitivity of glioblastoma multiforme. Int. J. Radiat. Oncol. 1995, 32, 99–104. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [Green Version]
- Skripcak, T.; Belka, C.; Bosch, W.; Brink, C.; Brunner, T.; Budach, V.; Büttner, D.; Debus, J.; Dekker, A.; Grau, C.; et al. Creating a data exchange strategy for radiotherapy research: Towards federated databases and anonymised public datasets. Radiother. Oncol. 2014, 113, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.T.; Finkel, K.A.; Warner, K.A.; Nör, F.; Tice, D.; Martins, M.D.; Jackson, T.L.; Nör, J.E. Patient-derived xenograft (PDX) tumors increase growth rate with time. Oncotarget 2016, 7, 7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.L.; Pokorny, J.L.; Schroeder, M.A.; Sarkaria, J.N. Establishment, maintenance, and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. In Current Protocols in Pharmacology; Enna, S.J., Williams, M., Barret, J.F., Ferkany, J.W., Kenakin, T., Porsolt, R.D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; ISBN 978-0-471-14175-4. [Google Scholar]
- William, D.; Mullins, C.S.; Schneider, B.; Orthmann, A.; Lamp, N.; Krohn, M.; Hoffmann, A.; Classen, C.-F.; Linnebacher, M. Optimized creation of glioblastoma patient derived xenografts for use in preclinical studies. J. Transl. Med. 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccirillo, S.G.M.; Combi, R.; Cajola, L.; Patrizi, A.; Redaelli, S.; Bentivegna, A.; Baronchelli, S.; Maira, G.; Pollo, B.; Mangiola, A.; et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene 2009, 28, 1807–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, L.; Angioi-Duprez, K.S.; Bracard, S.R.; Klein-Monhoven, N.A.; Le Faou, A.E.; Duprez, A.M.; Plénat, F.M. Analysis of tissue chimerism in nude mouse brain and abdominal xenograft models of human glioblastoma multiforme: What does it tell us about the models and about glioblastoma biology and therapy? J. Histochem. Cytochem. 2000, 48, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; Anderson, W.C.; Santaguida, M.T.; Dylla, S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Investig. 2013, 93, 970. [Google Scholar] [CrossRef]
- Jung, J.; Seol, H.S.; Chang, S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2018, 50, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Ivanics, T.; Bergquist, J.R.; Liu, G.; Kim, M.P.; Kang, Y.; Katz, M.H.; Perez, M.V.R.; Thomas, R.M.; Fleming, J.B.; Truty, M.J. Patient-derived xenograft cryopreservation and reanimation outcomes are dependent on cryoprotectant type. Lab. Investig. J. Tech. Methods Pathol. 2018, 98, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Guerrera, F.; Tabbò, F.; Bessone, L.; Maletta, F.; Gaudiano, M.; Ercole, E.; Annaratone, L.; Todaro, M.; Boita, M.; Filosso, P.L.; et al. The influence of tissue ischemia time on RNA integrity and patient-derived xenografts (PDX) engraftment rate in a non-small cell lung cancer (NSCLC) biobank. PLoS ONE 2016, 11, e0145100. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, K.; Ohara, M.; Saito, K.; Ozaki, S.; Terai, M.; Mastrangelo, M.J.; Fortina, P.; Aplin, A.E.; Sato, T. Establishment of an orthotopic patient-derived xenograft mouse model using uveal melanoma hepatic metastasis. J. Transl. Med. 2017, 15, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.; Temme, A.; Senner, V.; Ebner, R.; Schwind, S.; Stevanovic, S.; Wehner, R.; Schackert, G.; Schackert, H.K.; Fussel, M.; et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br. J. Cancer 2007, 96, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 Promotes Malignancy in Glioblastoma by Regulating Plasticity and Astrocytic Differentiation. Neoplasia 2014, 16, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Strojnik, T.; Røsland, G.V.; Sakariassen, P.O.; Kavalar, R.; Lah, T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: Correlation of nestin with prognosis of patient survival. Surg. Neurol. 2007, 68, 133–143. [Google Scholar] [CrossRef]
- Lin, A. Role of nestin in glioma invasion. World J. Transl. Med. 2015, 4, 78. [Google Scholar] [CrossRef]
- Lv, D.; Lu, L.; Hu, Z.; Fei, Z.; Liu, M.; Wei, L.; Xu, J. Nestin expression is associated with poor clinicopathological features and prognosis in glioma patients: An association study and meta-analysis. Mol. Neurobiol. 2017, 54, 727–735. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Wang, M.; Rojiani, A.M.; Tofilon, P.J.; Chakravarti, A.; Ang, K.K.; Zhang, H.-Z.; Hammond, E.; Curran, W.; Mehta, M.P. The prognostic value of nestin expression in newly diagnosed glioblastoma: Report from the Radiation Therapy Oncology Group. Radiat. Oncol. Lond. Engl. 2008, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.J.; Lan, F.; He, Q.; Lee, A.; Tang, C.Z.; Dong, L.; Lan, B.; Ma, X.; Wu, J.C.; Shen, L. Inducible expression of stem cell associated intermediate filament nestin reveals an important role in glioblastoma carcinogenesis. Int. J. Cancer 2011, 128, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.M.; Riedlinger, G.M.; Rosenfeld, J.; Ganesan, S.; Pine, S.R. Patient-derived xenograft models of non-small cell lung cancer and their potential utility in personalized medicine. Front. Oncol. 2017, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood–brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yaromina, A.; Krause, M.; Thames, H.; Rosner, A.; Krause, M.; Hessel, F.; Grenman, R.; Zips, D.; Baumann, M. Pre-treatment number of clonogenic cells and their radiosensitivity are major determinants of local tumour control after fractionated irradiation. Radiother. Oncol. 2007, 83, 304–310. [Google Scholar] [CrossRef]
- Gurtner, K.; Hessel, F.; Eicheler, W.; Dörfler, A.; Zips, D.; Heider, K.-H.; Krause, M.; Baumann, M. Combined treatment of the immunoconjugate bivatuzumab mertansine and fractionated irradiation improves local tumour control in vivo. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2012, 102, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, I.; Rolff, J.; Soong, R.; Hoffmann, J.; Hammer, S.; Sommer, A.; Becker, M.; Merk, J. Establishment of patient-derived non–small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin. Cancer Res. 2008, 14, 6456–6468. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.G.; Yu, M.; Spratt, D.E.; Chang, S.L.; Feng, F.Y.; Kim, M.M.; Speers, C.W.; Carlson, B.L.; Mladek, A.C.; Lawrence, T.S.; et al. Xenograft-based, platform-independent gene signatures to predict response to alkylating chemotherapy, radiation, and combination therapy for glioblastoma. Neuro Oncol. 2019, 21, 1141–1149. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Wang, G.; Lin, Y.-C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo murine-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Creighton, C.J.; Zhang, Y.; Sen, B.; Mazumdar, T.; Myers, J.N.; Woolfson, A.; Lorenzi, M.V.; Bell, D.; Williams, M.D.; et al. Tumor grafts derived from patients with head and neck squamous carcinoma authentically maintain the molecular and histologic characteristics of human cancers. J. Transl. Med. 2013, 11, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and phenotypic characterization of a broad panel of patient derived xenografts reflects the diversity of glioblastoma. Clin. Cancer Res. 2019, 26, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A Renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braekeveldt, N.; Von Stedingk, K.; Fransson, S.; Martinez-Monleon, A.; Lindgren, D.; Axelson, H.; Levander, F.; Willforss, J.; Hansson, K.; Øra, I.; et al. Patient-derived xenograft models reveal intratumor heterogeneity and temporal stability in neuroblastoma. Cancer Res. 2018, 78, 5958–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 2016, 167, 260–274. [Google Scholar] [CrossRef] [Green Version]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Smiraglia, D.J.; Rush, L.J.; Frühwald, M.C.; Dai, Z.; Held, W.A.; Costello, J.F.; Lang, J.C.; Eng, C.; Li, B.; Wright, F.A.; et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum. Mol. Genet. 2001, 10, 1413–1419. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Vijayachandra, K.; Penner, C.; Glick, A. Increased sensitivity of transforming growth factor (TGF) β1 null cells to alkylating agents reveals a novel link between TGFβ signaling and O 6-methylguanine methyltransferase promoter hypermethylation. J. Biol. Chem. 2001, 276, 19052–19058. [Google Scholar] [CrossRef] [Green Version]
- Danam, R.P.; Howell, S.R.; Remack, J.S.; Brent, T.P. Heterogeneous methylation of the O6-methylguanine-DNA methyltransferase promoter in immortalized IMR90 cell lines. Int. J. Oncol. 2001, 18, 1187–1193. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Christians, A.; Hartmann, C.; Benner, A.; Meyer, J.; von Deimling, A.; Weller, M.; Wick, W.; Weiler, M. Prognostic value of three different methods of mgmt promoter methylation analysis in a prospective trial on newly diagnosed glioblastoma. PLoS ONE 2012, 7, e33449. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort ID | Gender | Age | Methylation Status | Tumour Location |
|---|---|---|---|---|
| DK26 | f | 67 | methylated | frontal |
| DK27 | f | 72 | methylated | frontal |
| DK28 | f | 74 | unmethylated | parieto-occipital |
| DK29 | f | 66 | unmethylated | multi-ocular/fronto-parietal |
| DK30 | m | 57 | methylated | opercular |
| DK32 | m | 66 | unmethylated | parieto-occipital |
| DK33 | f | 73 | methylated | frontal-temporal |
| DK34 | f | 82 | methylated | parieto-occipital |
| DK35 | f | 75 | unmethylated | parieto-occipital |
| DK36 | m | 85 | unmethylated | frontal |
| DK37 | m | 79 | unmethylated | temporal lobe |
| DK38 | m | 73 | unmethylated | frontal |
| DK39 | m | 63 | methylated | parieto-temporal to occipital |
| DK40 | f | 74 | methylated | frontal lobe |
| DK41 | m | 53 | unmethylated | frontal lobe |
| DK42 | m | 71 | unmethylated | frontal lobe |
| DK45 | m | 79 | unmethylated | temporal-occipital |
| DK51 | m | 67 | methylated | frontal lobe |
| DK54 | f | 78 | unmethylated | temporo-parietal |
| DK60 | m | 76 | unmethylated | temporo-parietal |
| DK63 | f | 76 | methylated | frontal |
| DK77 | f | 83 | methylated | temporal-occipital |
| Experiment | Take Proportion | Number of Passages |
|---|---|---|
| DK26 | 7/10 | 3 |
| DK28 | 2/10 | 1 |
| DK29 | 7/10 | 5 |
| DK30 | 2/10 | 2 |
| DK32 | 1/10 | 1 |
| DK33 | 7/10 | 3 |
| DK35 | 1/5 | 2 |
| DK39 | 5/10 | 3 |
| DK41 | 5/10 | 1 |
| DK42 | 3/10 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meneceur, S.; Linge, A.; Meinhardt, M.; Hering, S.; Löck, S.; Bütof, R.; Krex, D.; Schackert, G.; Temme, A.; Baumann, M.; et al. Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma. Cancers 2020, 12, 871. https://doi.org/10.3390/cancers12040871
Meneceur S, Linge A, Meinhardt M, Hering S, Löck S, Bütof R, Krex D, Schackert G, Temme A, Baumann M, et al. Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma. Cancers. 2020; 12(4):871. https://doi.org/10.3390/cancers12040871
Chicago/Turabian StyleMeneceur, Sarah, Annett Linge, Matthias Meinhardt, Sandra Hering, Steffen Löck, Rebecca Bütof, Dietmar Krex, Gabriele Schackert, Achim Temme, Michael Baumann, and et al. 2020. "Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma" Cancers 12, no. 4: 871. https://doi.org/10.3390/cancers12040871
APA StyleMeneceur, S., Linge, A., Meinhardt, M., Hering, S., Löck, S., Bütof, R., Krex, D., Schackert, G., Temme, A., Baumann, M., Krause, M., & von Neubeck, C. (2020). Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma. Cancers, 12(4), 871. https://doi.org/10.3390/cancers12040871