The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
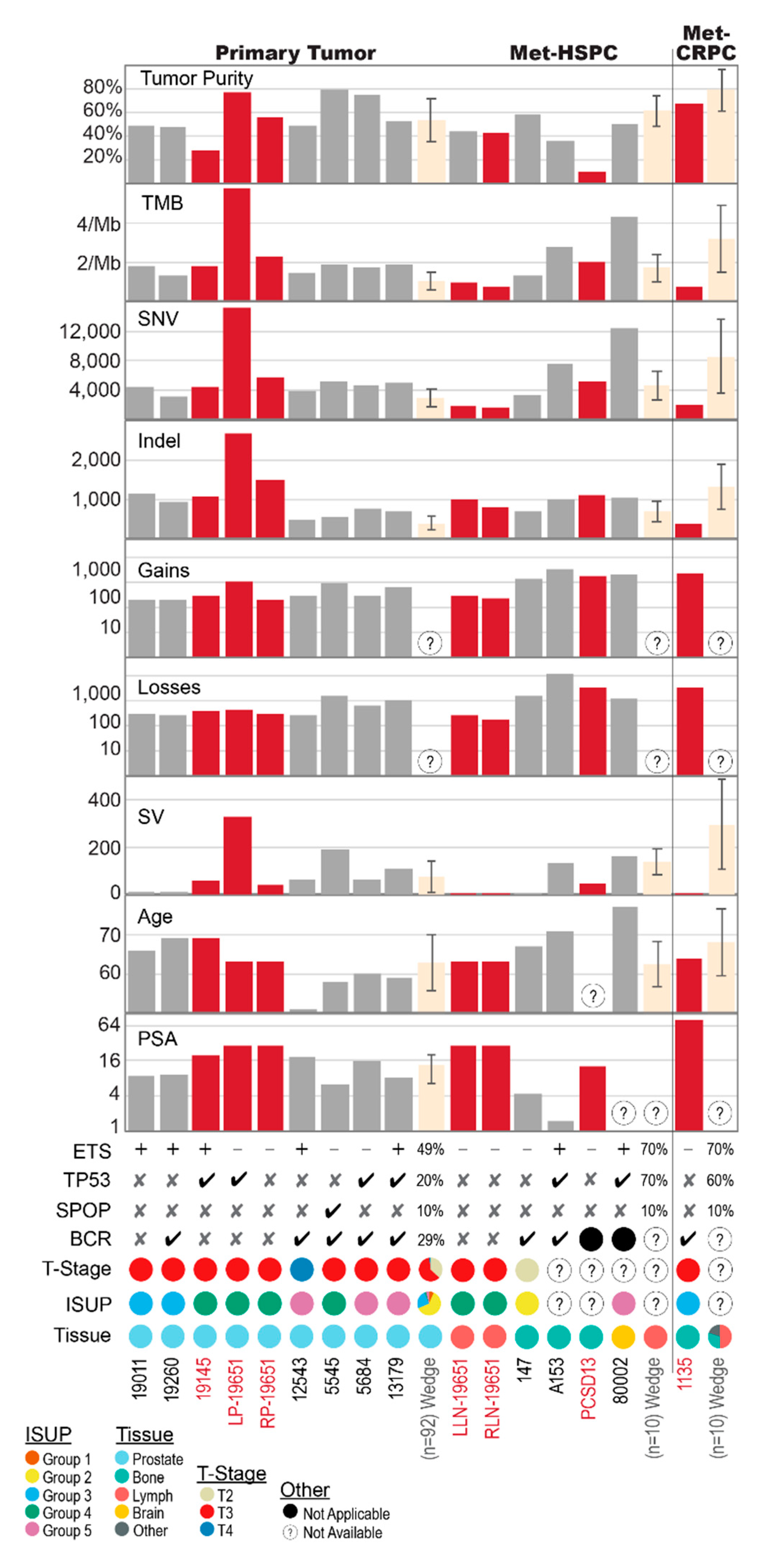
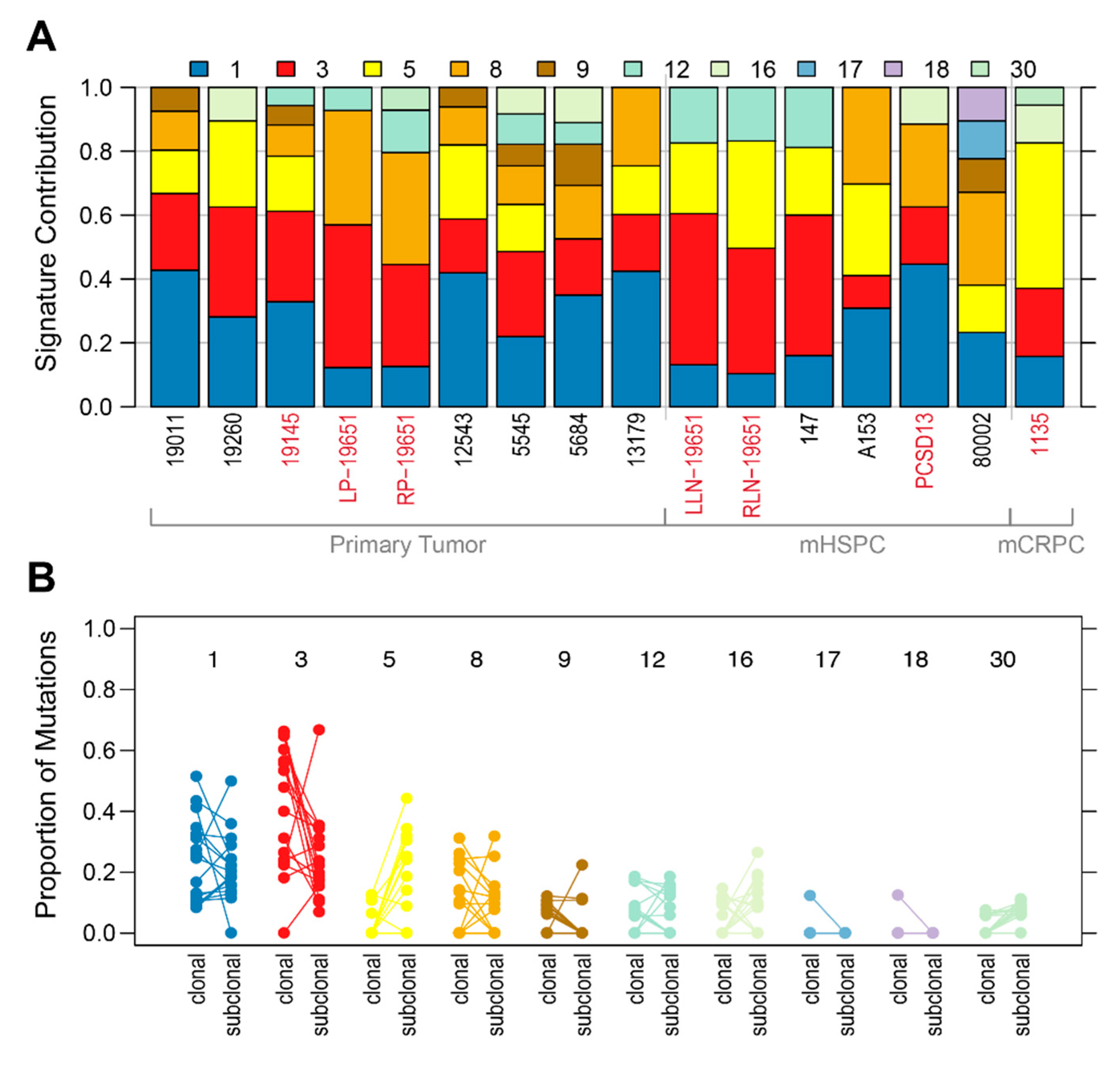
2.1. Shared Genomic Landscape
2.2. Primary Prostate Samples with Synchronous Lymph Node Metastases: 19011, 19260, 19145 and 19651
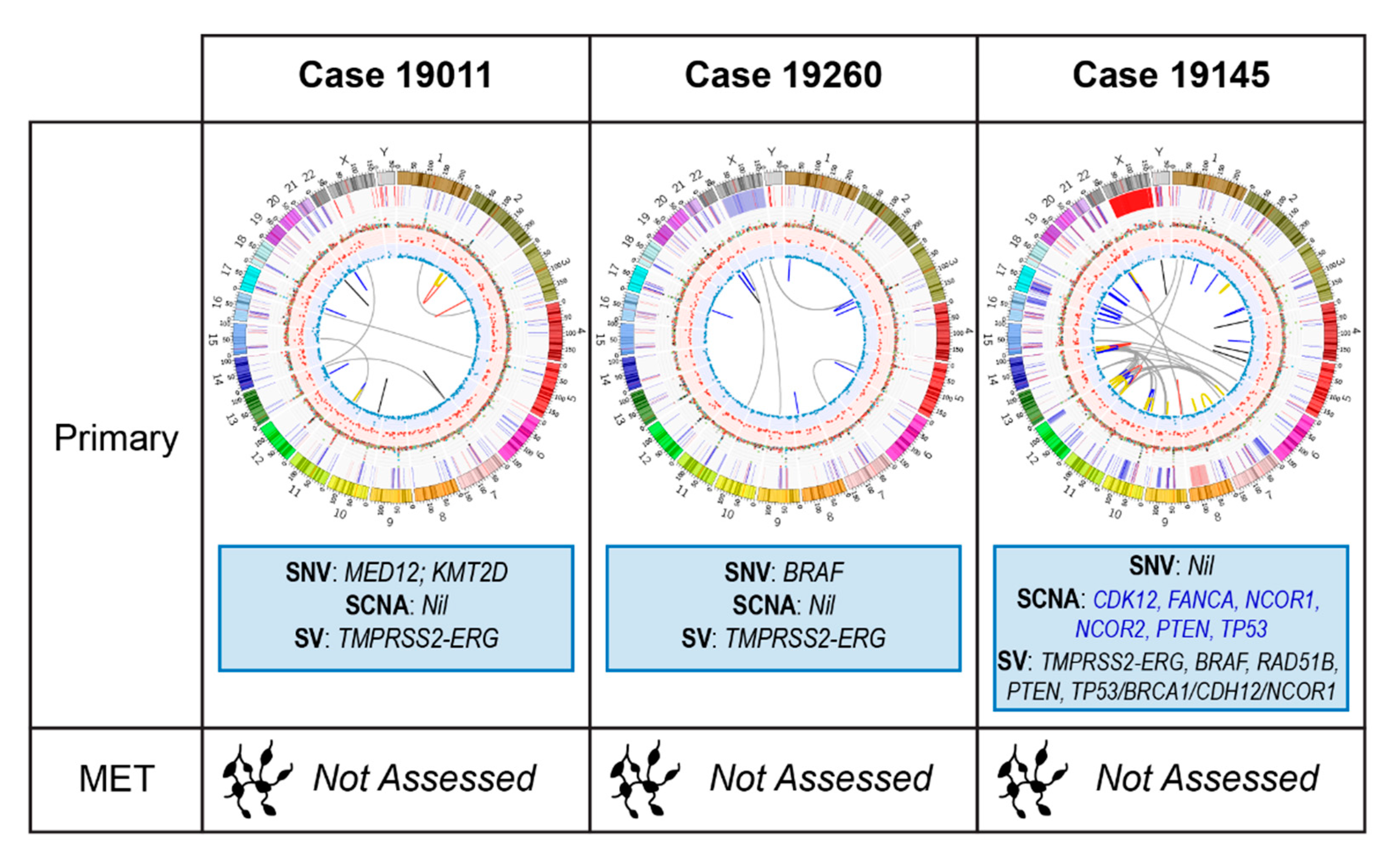
2.2.1. Case 19011: Left Prostate Tumour Core Biopsy
2.2.2. Case 19260: Right Prostate Tumour Core Biopsy
2.2.3. Case 19145: Left Prostate Tumour Core Biopsy
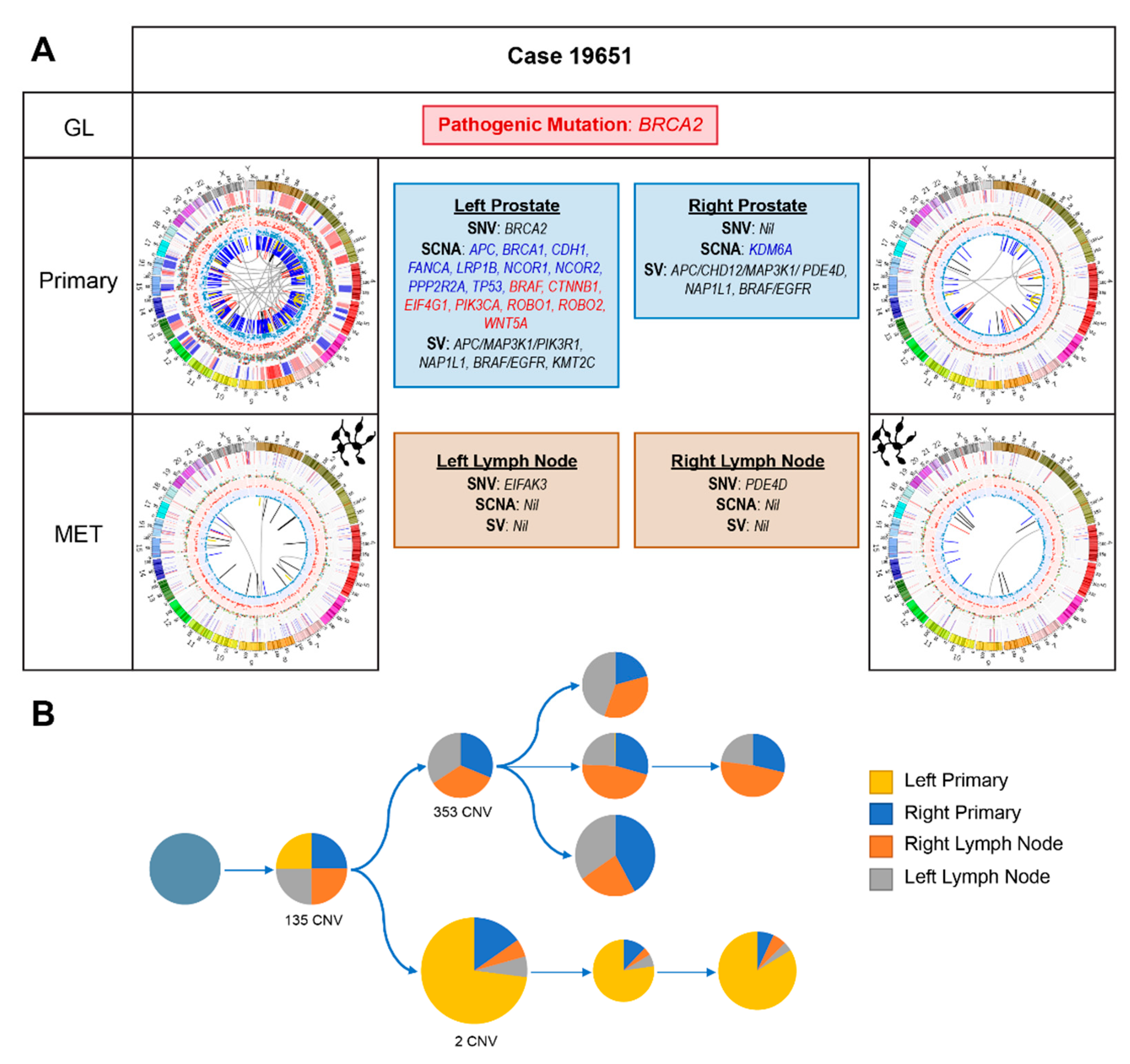
2.2.4. Case 19651: Bilateral Prostate and Internal Iliac Node Tumour Core Biopsies
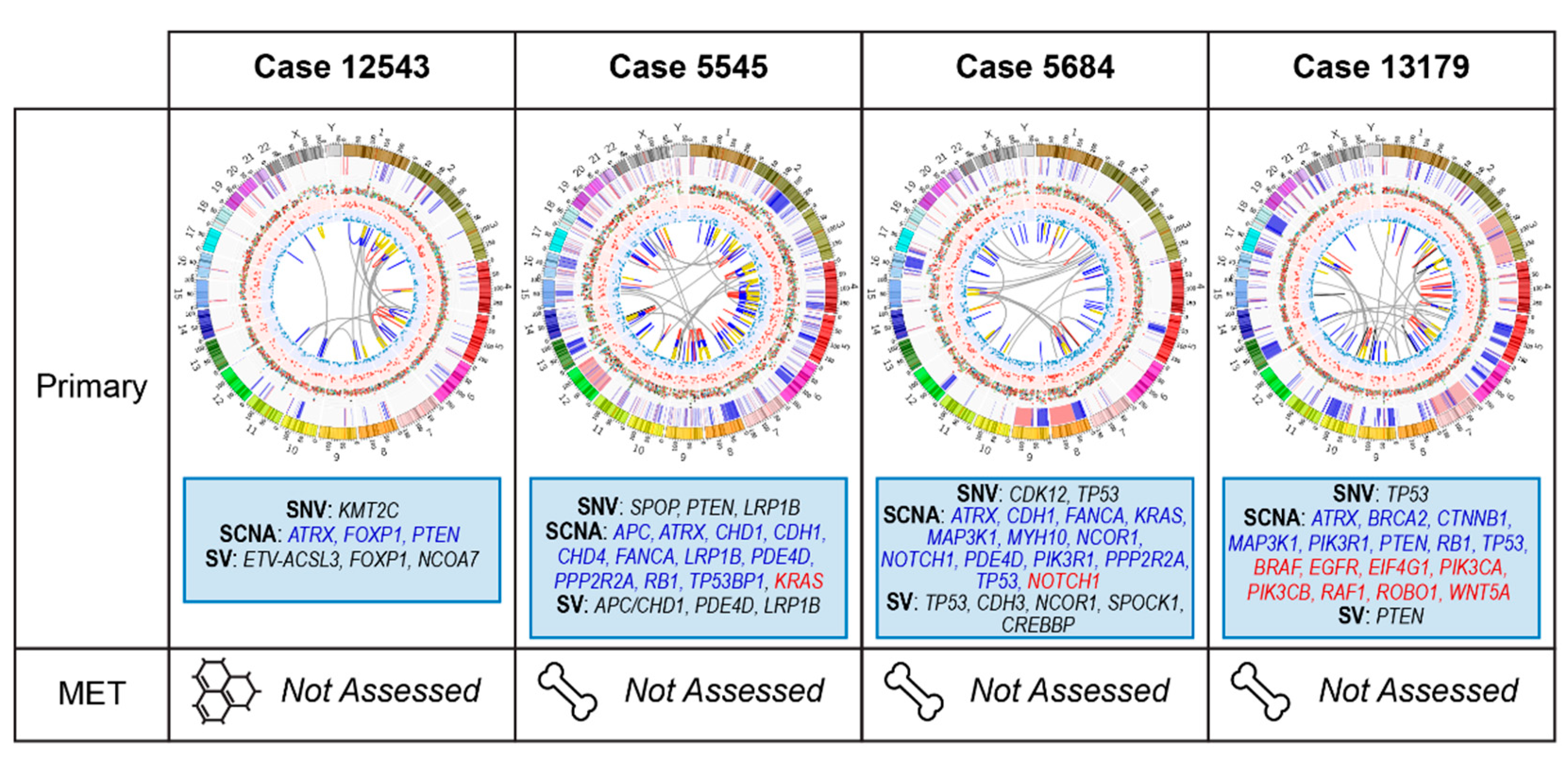
2.3. Primary Prostate Samples with Relapse Post Radical Prostatectomy: 12543, 5545, 5684, and 13179
2.3.1. Case 12543: Left Prostate Tumour Core Biopsy
2.3.2. Case 5545: Left Prostate Tumour Core Biopsy
2.3.3. Case 5684: Right Prostate Tumour Core Biopsy
2.3.4. Case 13179: Right Prostate Tumour Core Biopsy
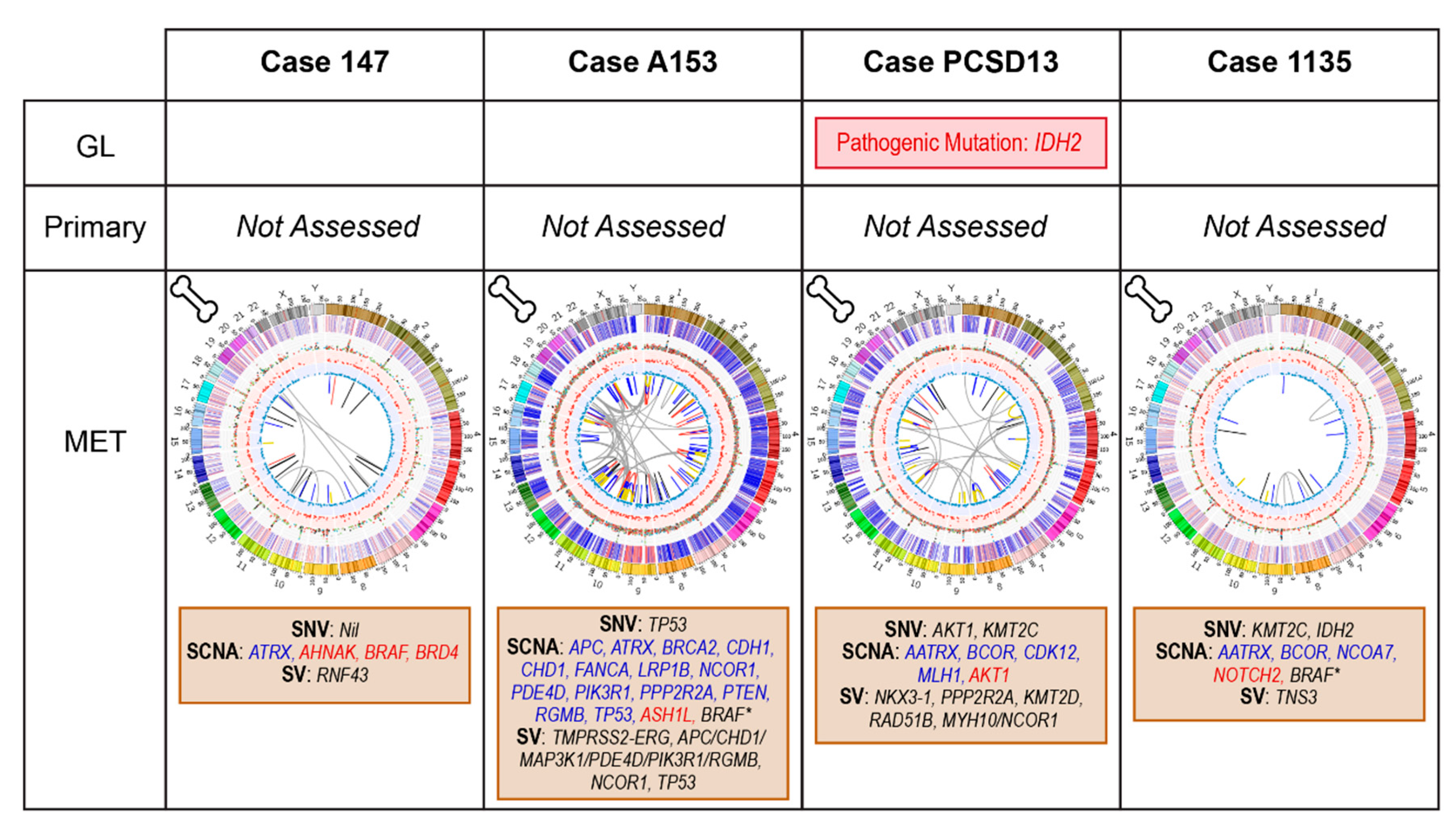
2.4. Bone Metastatic Samples: 147, A153, PCSD13 and 1135
2.4.1. Case 147: Biopsy Left Pubic Bone Corresponding to Sclerotic Region on Imaging
2.4.2. Case A153: Biopsy Right Iliac Crest Corresponding to Metastatic Deposit on Imaging
2.4.3. Case PCSD13: Biopsy Left Femur during Total Hip Replacement for Pathological Fracture
2.4.4. Case 1135: Biopsy Right Posterior Iliac Crest Corresponding to Metastatic Deposit on Imaging
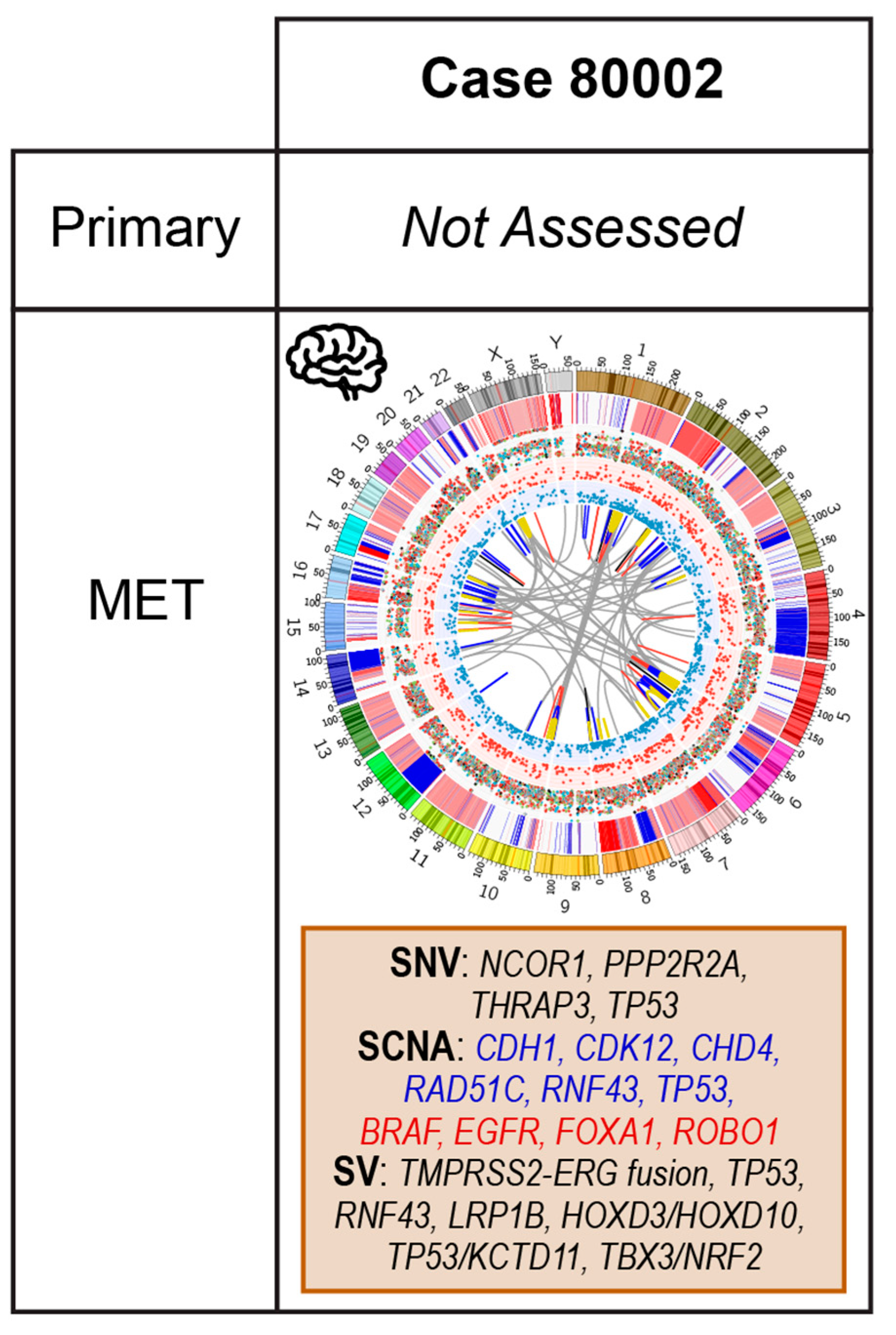
2.5. Case 80002: Core Biopsy at Resection of Brain Metastasis
3. Materials and Methods
3.1. Whole Genome Sequencing (WGS)
3.2. WGS Variant Calling
3.3. WGS Variant Annotation
3.4. Tumour Mutational Burden and Percentage Genome Alteration
3.5. Whole Genome Optical Mapping
3.6. WGM Derived Genomic Rearrangements
3.7. WGM Derived Data Filtering
3.8. Generation of a Prostate Cancer-Related Gene List
3.9. Other Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, S.A.; Hu, C.; Sartor, O.; Gomella, L.G.; Amin, M.B.; Purdy, J.; Michalski, J.M.; Garzotto, M.G.; Pervez, N.; Balogh, A.G.; et al. Effect of Chemotherapy With Docetaxel With Androgen Suppression and Radiotherapy for Localized High-Risk Prostate Cancer: The Randomized Phase III NRG Oncology RTOG 0521 Trial. J. Clin. Oncol. 2019, 37, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.C.; James, N.D.; Brawley, C.D.; Clarke, N.W.; Hoyle, A.P.; Ali, A.; Ritchie, A.W.S.; Attard, G.; Chowdhury, S.; Cross, W.; et al. Systemic Therapy for Advanced or Metastatic Prostate cancer: Evaluation of Drug Efficacy (STAMPEDE) investigators Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): A randomised controlled phase 3 trial. Lancet 2018, 392, 2353–2366. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Clark, J.; Ambroisine, L.; Mills, I.G.; Fisher, G.; Flohr, P.; Reid, A.; Edwards, S.; Kovacs, G.; Berney, D.; et al. Transatlantic Prostate Group Heterogeneity and clinical significance of ETV1 translocations in human prostate cancer. Br. J. Cancer 2008, 99, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansen, A.; Bergström, R.; Delahunt, B.; Samaratunga, H.; Guðjónsdóttir, J.; Grönberg, H.; Egevad, L.; Lindberg, J. Somatic alterations detected in diagnostic prostate biopsies provide an inadequate representation of multifocal prostate cancer. Prostate 2019, 79, 920–928. [Google Scholar] [CrossRef]
- Hotte, S.J.; Joshua, A.M.; Torria, V.; Macfarlane, R.J.; Basappa, N.S.; Winquist, J.P.; Mukherjee, S.; Gregg, R.W.; Kollmannsberger, C.K.; Finch, D.L.; et al. NCIC CTG, IND-205: A phase II study of PX-866 in patients with recurrent or metastatic castration-resistant prostate cancer (CRPC). J. Clin. Oncol. 2013, 31, 5042. [Google Scholar] [CrossRef]
- Hong, D.S.; Bowles, D.W.; Falchook, G.S.; Messersmith, W.A.; George, G.C.; O’Bryant, C.L.; Vo, A.C.H.; Klucher, K.; Herbst, R.S.; Eckhardt, S.G.; et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 4173–4182. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, S.K.; Omlin, A.; Hylands, L.; Miranda, S.; Barber, L.J.; Riisnaes, R.; Reid, A.H.; Attard, G.; Chen, L.; Kozarewa, I.; et al. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer. Ann. Oncol. 2013, 24, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudmundsson, J.; Sulem, P.; Rafnar, T.; Bergthorsson, J.T.; Manolescu, A.; Gudbjartsson, D.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsdottir, K.R.; Blondal, T.; et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat. Genet. 2008, 40, 281–283. [Google Scholar] [CrossRef] [Green Version]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Müller, S.; Gärtner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002, 62, 840–847. [Google Scholar]
- Zhang, X.; Cowper-Sal lari, R.; Bailey, S.D.; Moore, J.H.; Lupien, M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res. 2012, 22, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef]
- Lang, S.H.; Swift, S.L.; White, H.; Misso, K.; Kleijnen, J.; Quek, R.G.W. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int. J. Oncol. 2019, 55, 597–616. [Google Scholar] [CrossRef] [Green Version]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Isaacsson Velho, P.; Fu, W.; Wang, H.; Mirkheshti, N.; Qazi, F.; Lima, F.A.S.; Shaukat, F.; Carducci, M.A.; Denmeade, S.R.; Paller, C.J.; et al. Wnt-pathway Activating Mutations Are Associated with Resistance to First-line Abiraterone and Enzalutamide in Castration-resistant Prostate Cancer. Eur. Urol. 2020, 77, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Morova, T.; McNeill, D.R.; Lallous, N.; Gönen, M.; Dalal, K.; Wilson, D.M.; Gürsoy, A.; Keskin, Ö.; Lack, N.A. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Hawley, J.R.; Soares, F.; Grillo, G.; Teng, M.; Madani Tonekaboni, S.A.; Hua, J.T.; Kron, K.J.; Mazrooei, P.; Ahmed, M.; et al. Noncoding mutations target cis-regulatory elements of the FOXA1 plexus in prostate cancer. Nat. Commun. 2020, 11, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 2018, 174, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, M.; Orr, N.; Hayes, R.B.; Jacobs, K.B.; Kraft, P.; Wacholder, S.; Minichiello, M.J.; Fearnhead, P.; Yu, K.; Chatterjee, N.; et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 2007, 39, 645–649. [Google Scholar] [CrossRef]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-C.; Hu, C.-D. Transcriptional activity of c-Jun is critical for the suppression of AR function. Mol. Cell. Endocrinol. 2013, 372, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Eggener, S.E.; Scardino, P.T.; Walsh, P.C.; Han, M.; Partin, A.W.; Trock, B.J.; Feng, Z.; Wood, D.P.; Eastham, J.A.; Yossepowitch, O.; et al. Predicting 15-year prostate cancer specific mortality after radical prostatectomy. J. Urol. 2011, 185, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Touijer, K.A.; Karnes, R.J.; Passoni, N.; Sjoberg, D.D.; Assel, M.; Fossati, N.; Gandaglia, G.; Eastham, J.A.; Scardino, P.T.; Vickers, A.; et al. Survival Outcomes of Men with Lymph Node-positive Prostate Cancer After Radical Prostatectomy: A Comparative Analysis of Different Postoperative Management Strategies. Eur. Urol. 2018, 73, 890–896. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. STAMPEDE Investigators Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- James, N.; Woods, B.; Sideris, E.; Spears, M.R.; Dearnaley, D.P.; Mason, M.; Clarke, N.; Parmar, M.K.B.; Sydes, M.R.; Sculpher, M. STAMPEDE Investigators Addition of docetaxel to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Long-term survival, quality-adjusted survival, and cost-effectiveness analysis. J. Clin. Oncol. 2018, 36, 162. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, M.D.; Harris, J.; Sartor, O.; Xiao, Y.; Shayegan, B.; Sperduto, P.W.; Badiozamani, K.R.; Lawton, C.A.F.; Horwitz, E.M.; Michalski, J.M.; et al. Adjuvant radiation therapy, androgen deprivation, and docetaxel for high-risk prostate cancer postprostatectomy: Results of NRG Oncology/RTOG study 0621. Cancer 2017, 123, 2489–2496. [Google Scholar] [CrossRef]
- Eastham, J.A.; Heller, G.; Halabi, S.; Monk, P.; Clinton, S.K.; Szmulewitz, R.Z.; Coleman, J.; Gleave, M.; Evans, C.P.; Hillman, D.W.; et al. CALGB 90203 (Alliance): Radical prostatectomy (RP) with or without neoadjuvant chemohormonal therapy (CHT) in men with clinically localized, high-risk prostate cancer (CLHRPC). J. Clin. Oncol. 2019, 37, 5079. [Google Scholar] [CrossRef]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann. Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Prophet, M.; Xiao, K.; Gourdin, T.S.; Nagy, R.J.; Kiedrowski, L.A.; Ledet, E.; Sonpavde, G.; Sartor, A.O.; Lilly, M.B. Detection of actionableBRAF missense mutations by ctDNA-based genomic analysis in prostate cancer. J. Clin. Oncol. 2018, 36, 306. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Forster, M.D.; Dedes, K.J.; Sandhu, S.; Frentzas, S.; Kristeleit, R.; Ashworth, A.; Poole, C.J.; Weigelt, B.; Kaye, S.B.; Molife, L.R. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 302–306. [Google Scholar] [CrossRef]
- Wang, J. Complete pathological remission after treatment with olaparib in a patient with PTEN-deficient sarcomatoid prostate cancer. J. Mol. Cancer 2018, 1, 17–19. [Google Scholar]
- González-Billalabeitia, E.; Seitzer, N.; Song, S.J.; Song, M.S.; Patnaik, A.; Liu, X.-S.; Epping, M.T.; Papa, A.; Hobbs, R.M.; Chen, M.; et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014, 4, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, M.C.; Astapova, I.; Cheng, S.; Lee, L.J.; Verhoeven, M.C.; Choi, E.; Balk, S.P.; Hollenberg, A.N. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J. Biol. Chem. 2005, 280, 6511–6519. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Yang, Y.; Zhang, C.; Hu, P.; Kan, W.; Bai, X.; Liu, X.; Song, H. FBI-1 functions as a novel AR co-repressor in prostate cancer cells. Cell Mol. Life Sci. 2011, 68, 1091–1103. [Google Scholar] [CrossRef]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear receptor corepressor complexes in cancer: Mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar]
- Lopez, S.M.; Agoulnik, A.I.; Zhang, M.; Peterson, L.E.; Suarez, E.; Gandarillas, G.A.; Frolov, A.; Li, R.; Rajapakshe, K.; Coarfa, C.; et al. Nuclear Receptor Corepressor 1 Expression and Output Declines with Prostate Cancer Progression. Clin. Cancer Res. 2016, 22, 3937–3949. [Google Scholar] [CrossRef] [Green Version]
- Lorenzin, F.; Demichelis, F. Evolution of the prostate cancer genome towards resistance. JTGG J. Transl. Genet. Genom. 2019, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Maughan, B.L.; Guedes, L.B.; Boucher, K.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Antonarakis, E.S.; Lotan, T.L. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018, 173, 1770–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Løvf, M.; Zhao, S.; Axcrona, U.; Johannessen, B.; Bakken, A.C.; Carm, K.T.; Hoff, A.M.; Myklebost, O.; Meza-Zepeda, L.A.; Lie, A.K.; et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur. Urol. 2019, 75, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.M.; Evans, D.G.R.; Hope, Q.; Norman, A.R.; Barbachano, Y.; Bullock, S.; Kote-Jarai, Z.; Meitz, J.; Falconer, A.; Osin, P.; et al. UK Genetic Prostate Cancer Study Collaborators and BAUS Section of Oncology Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br. J. Cancer 2010, 103, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and are Associated with Early Age at Death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef] [Green Version]
- Annala, M.; Struss, W.J.; Warner, E.W.; Beja, K.; Vandekerkhove, G.; Wong, A.; Khalaf, D.; Seppälä, I.-L.; So, A.; Lo, G.; et al. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur. Urol. 2017, 72, 34–42. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Spisák, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O’Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef] [Green Version]
- Oudard, S.; Latorzeff, I.; Caty, A.; Miglianico, L.; Sevin, E.; Hardy-Bessard, A.C.; Delva, R.; Rolland, F.; Mouret, L.; Priou, F.; et al. Effect of Adding Docetaxel to Androgen-Deprivation Therapy in Patients With High-Risk Prostate Cancer With Rising Prostate-Specific Antigen Levels After Primary Local Therapy: A Randomized Clinical Trial. JAMA Oncol. 2019, 5, 623–632. [Google Scholar] [CrossRef]
- Migita, T.; Takayama, K.-I.; Urano, T.; Obinata, D.; Ikeda, K.; Soga, T.; Takahashi, S.; Inoue, S. ACSL3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci. 2017, 108, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Hieronymus, H.; Iaquinta, P.J.; Wongvipat, J.; Gopalan, A.; Murali, R.; Mao, N.; Carver, B.S.; Sawyers, C.L. Deletion of 3p13-14 locus spanning FOXP1 to SHQ1 cooperates with PTEN loss in prostate oncogenesis. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desotelle, J.; Truong, M.; Ewald, J.; Weeratunga, P.; Yang, B.; Huang, W.; Jarrard, D. CpG island hypermethylation frequently silences FILIP1L isoform 2 expression in prostate cancer. J. Urol. 2013, 189, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Sumanasuriya, S.; Bianchini, D.; et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef]
- Tucker, M.D.; Zhu, J.; Marin, D.; Gupta, R.T.; Gupta, S.; Berry, W.R.; Ramalingam, S.; Zhang, T.; Harrison, M.; Wu, Y.; et al. Pembrolizumab in men with heavily treated metastatic castrate-resistant prostate cancer. Cancer Med. 2019, 8, 4644–4655. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: Stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol. 1998, 18, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F.A.; et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef]
- Chen, Q.; Yao, Y.-T.; Xu, H.; Chen, Y.-B.; Gu, M.; Cai, Z.-K.; Wang, Z. SPOCK1 promotes tumor growth and metastasis in human prostate cancer. Drug Des. Dev. Ther. 2016, 10, 2311–2321. [Google Scholar] [PubMed] [Green Version]
- Comuzzi, B.; Nemes, C.; Schmidt, S.; Jasarevic, Z.; Lodde, M.; Pycha, A.; Bartsch, G.; Offner, F.; Culig, Z.; Hobisch, A. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J. Pathol. 2004, 204, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Shafran, J.S.; Andrieu, G.P.; Györffy, B.; Denis, G.V. BRD4 Regulates Metastatic Potential of Castration-Resistant Prostate Cancer through AHNAK. Mol. Cancer Res. 2019, 17, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Li, X.; Baek, G.; Ramanand, S.G.; Sharp, A.; Gao, Y.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 2018, 22, 796–808. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019. [Google Scholar]
- Kotredes, K.P.; Razmpour, R.; Lutton, E.; Alfonso-Prieto, M.; Ramirez, S.H.; Gamero, A.M. Characterization of cancer-associated IDH2 mutations that differ in tumorigenicity, chemosensitivity and 2-hydroxyglutarate production. Oncotarget 2019, 10, 2675–2692. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Li, G.; Vass, W.C.; Papageorge, A.; Walker, R.C.; Asnaghi, L.; Steinbach, P.J.; Tosato, G.; Hunter, K.; Lowy, D.R. The Tensin-3 protein, including its SH2 domain, is phosphorylated by Src and contributes to tumorigenesis and metastasis. Cancer Cell 2009, 16, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Jiang, H.; Su, X.-M.; Zheng, L.; Li, Q.-L.; Zhang, Z.-W.; Li, X.-C. Expressions of E-cadherin and alpha-catenin in benign, malignant and metastatic prostate tumors. Zhonghua Nan Ke Xue 2012, 18, 499–503. [Google Scholar] [PubMed]
- Chang, F.; Xing, P.; Song, F.; Du, X.; Wang, G.; Chen, K.; Yang, J. The role of T-box genes in the tumorigenesis and progression of cancer. Oncol. Lett. 2016, 12, 4305–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, D.; Zhou, C.; Shi, Y.; Lu, H.; Xu, R.; He, X. Nuclear transcription factor Nrf2 suppresses prostate cancer cells growth and migration through upregulating ferroportin. Oncotarget 2016, 7, 78804–78812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [Green Version]
- Mo, R.J.; Lu, J.M.; Wan, Y.P.; Hua, W.; Liang, Y.X.; Zhuo, Y.J.; Kuang, Q.W.; Liu, Y.L.; He, H.C.; Zhong, W.D. Decreased hoxd10 expression promotes a proliferative and aggressive phenotype in prostate cancer. Curr. Mol. Med. 2017, 17, 70–78. [Google Scholar] [CrossRef]
- Kron, K.J.; Liu, L.; Pethe, V.V.; Demetrashvili, N.; Nesbitt, M.E.; Trachtenberg, J.; Ozcelik, H.; Fleshner, N.E.; Briollais, L.; van der Kwast, T.H.; et al. DNA methylation of HOXD3 as a marker of prostate cancer progression. Lab. Investig. 2010, 90, 1060–1067. [Google Scholar] [CrossRef]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assié, G.; André, T.; Hélias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.-F.; et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Tomkova, M.; Tomek, J.; Kriaucionis, S.; Schuster-Böckler, B. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tomkova, M.; Renard, C.; Urban, L.; Kolli, S.; Ardin, M.; Pandey, M.; Zhivagui, M.; Huskova, H.; Olivier, M.; Marusawa, H.; et al. Abstract 4661: Deciphering the causes of the COSMIC mutational signature 17 by combining pan-cancer data with experimental mouse models. In Tumor Biology; American Association for Cancer Research: Amherst, MA, USA, 2019; p. 4661. [Google Scholar]
- Hatzoglou, V.; Patel, G.V.; Morris, M.J.; Curtis, K.; Zhang, Z.; Shi, W.; Huse, J.; Rosenblum, M.; Holodny, A.I.; Young, R.J. Brain metastases from prostate cancer: An 11-year analysis in the MRI era with emphasis on imaging characteristics, incidence, and prognosis. J. Neuroimag. 2014, 24, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jin, H.; Zhao, J.C.; Yang, Y.A.; Li, Y.; Yang, X.; Dong, X.; Yu, J. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene 2017, 36, 4072–4080. [Google Scholar] [CrossRef] [Green Version]
- Zazzeroni, F.; Nicosia, D.; Tessitore, A.; Gallo, R.; Verzella, D.; Fischietti, M.; Vecchiotti, D.; Ventura, L.; Capece, D.; Gulino, A.; et al. KCTD11 tumor suppressor gene expression is reduced in prostate adenocarcinoma. Biomed Res. Int. 2014, 2014, 380398. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–11. [Google Scholar]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Cameron, D.L.; Schröder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P.; Papenfuss, A.T. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef] [Green Version]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, H.; Liang, H.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. CanDrA: Cancer-specific driver missense mutation annotation with optimized features. PLoS ONE 2013, 8, e77945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, J.S.; Fischer, B.; Lawrence, M.; Huber, W. SomaticSignatures: Inferring mutational signatures from single-nucleotide variants. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshwar, A.G.; Vembu, S.; Yung, C.K.; Jang, G.H.; Stein, L.; Morris, Q. PhyloWGS: Reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 2015, 16, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Ha, G.; Roth, A.; Khattra, J.; Ho, J.; Yap, D.; Prentice, L.M.; Melnyk, N.; McPherson, A.; Bashashati, A.; Laks, E.; et al. TITAN: Inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014, 24, 1881–1893. [Google Scholar] [CrossRef]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef]
- Hastie, A.R.; Dong, L.; Smith, A.; Finklestein, J.; Lam, E.T.; Huo, N.; Cao, H.; Kwok, P.-Y.; Deal, K.R.; Dvorak, J.; et al. Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLoS ONE 2013, 8, e55864. [Google Scholar] [CrossRef]
- Hastie, A.R.; Lam, E.T.; Pang, A.W.C.; Zhang, X.; Andrews, W.; Lee, J.; Liang, T.Y.; Wang, J.; Zhou, X.; Zhu, Z.; et al. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. BioRxiv 2017. [Google Scholar]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Bionano Data. Available online: https://protect-au.mimecast.com/s/QtA0COMK24uZZWrtEuAv2?domain=dx.doi.org (accessed on 2 March 2020).
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.; Lopez-Bigas, N.; et al. The repertoire of mutational signatures in human cancer. Nature. 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, J.; Bayard, Q.; Imbeaud, S.; Hirsch, T.Z.; Liu, F.; Renault, V.; Zucman-Rossi, J.; Letouzé, E. Palimpsest: An R package for studying mutational and structural variant signatures along clonal evolution in cancer. Bioinformatics 2018, 34, 3380–3381. [Google Scholar] [CrossRef] [PubMed]
- De Laere, B.; Oeyen, S.; Mayrhofer, M.; Whitington, T.; van Dam, P.-J.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 1766–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Time of Sample Collection for Genomic Interrogation | Relapse and Outcomes | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Age | Stage | Initial Treatment | ISUP | Clinical State | PSA | ECOG | Symptoms | Sample Site | ADT Prior | CRPC? | Time to BCR (mos) | Rx for BCR | Time to Mets (mos) | Duration Follow-Up (mos) |
| 1135 | 64 | T3N0 | RP | 3 | Rl | 80.7 | 1 | Yes | Bone | Yes | Yes | 33 | ADT | 68 | 68 |
| 19651 | 63 | T3N1 | ADT, RP, aRT | 4 | Dx | 28 | 0 | No | Prostate, nodes | Yes (6wks) | No | NR | NA | NR | 20 |
| 147 | 67 | T2N0 | RP | 2 | Rl | 4.4 | 0 | Yes | Bone | No | No | 84 | Nil | 107 | 135 |
| 19260 | 69 | T3N1 | RP | 3 | Dx | 9.2 | 0 | No | Prostate | No | No | 16 | sRT | NR | 27 |
| 5545 | 58 | T3N0 | RP | 4 | Dx | 6.3 | 0 | No | Prostate | No | No | 6 | sRT | 24 | 72 |
| 5684 | 60 | T3N0 | RP | 5 | Dx | 15.7 | 0 | No | Prostate | No | No | 36 | ADT | 120 | 132 |
| 19145 | 69 | T3N1 | RP, aRT | 4 | Dx | 20 | 0 | No | Prostate | Yes (4wks) | No | NR | NR | NA | 33 |
| 19011 | 66 | T3N1 | RP, aRT | 5 | Dx | 8.9 | 0 | No | Prostate | No | No | NR | NA | NA | 35 |
| 12543 | 51 | T4N0 | RP, aRT | 5 | Dx | 18.6 | 0 | No | Prostate | No | No | 72 | ADT | NA | 120 |
| 13179 | 59 | T3N0 | RP, aRT, ADT | 5 | Dx | 8.4 | 0 | No | Prostate | No | No | 51 | ADT | 51 | 51 |
| PCSD13 | 69 | TxNxM1 | ADT | - | Dx | 12.8 | 2 | Yes | Bone | Yes (8wks) | No | NA | NA | NA | 9 |
| A153 | 71 | T2N0 | RP | 3 | Rl | 1.5 | 0 | Yes | Bone | No | No | 93 | Nil | 105 | 120 |
| 80002 | 77 | TxNxM1 | Resection, ADT | 5 | Dx | - | 1 | Yes | Brain | No | No | NA | NA | NA | 1 |
| PI3K Pathway | MAPK Pathway | WNT Pathway | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Cases | Event | Gene | Cases | Event | Gene | Cases | Event |
| PTEN | 554512543, 1914519145 | SNVSCNASV | BRAF | 1926019651LP, 13179, 1135, 147, A153, 8000219145,19651LP+RP, 80002 | SNVSCNASV | APC | 19651LP, 5545, A15319651LP+RP, 12543, 5545, A153 | SCNASV |
| PIK3CA | 19651LP, 13179 | SCNA | EGFR | 13179, 80002 | SCNA | CTNNB1 | 19651LP, 13179 | SCNA |
| PIK3CB | 13179 | SCNA | KRAS | 19145, 5545, 5684 | SCNA | RNF43 | 80002, 14780002, 147 | SCNASV |
| PIK3R1 | 5684, 13179 | SCNA | MAP3K1 | 5684, 13179,A153, 19651RP+LP | SCNASV | WNT5A | 19651LP, 13179, 8000219260 | SCNASV |
| AKT1 | PCSD13 | SNV, SCNA | RAF1 | 19651LP, 13179, 147, 80002 | SCNA | MED12 | 19011 | Germline SNV |
| Disease State of Sample | Sample ID | SVs from Whole Genome Optical Mapping | % Missed by Whole Genome Sequencing | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | INS | DEL | DUP | INV | Intra-Chr | Inter-Chr | Total | INS | DEL | DUP | INV | Intra-Chr | Inter-Chr | ||
| Primary Tumor | 5545 | 39 | 0 | 27 | 3 | 0 | 7 | 2 | 36 | - | 37 | 67 | - | 29 | 0 |
| 13179 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 100 | - | 100 | - | - | - | - | |
| 5684 | 5 | 3 | 2 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | - | - | - | - | |
| 12543 | 10 | 1 | 5 | 0 | 0 | 1 | 3 | 60 | 0 | 40 | - | - | 100 | 67 | |
| 19651LP | 91 | 5 | 33 | 3 | 1 | 19 | 30 | 29 | 60 | 39 | 0 | 0 | 11 | 27 | |
| Met HSPC | A153 | 8 | 6 | 1 | 1 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | - | - | - |
| 147 | 5 | 4 | 0 | 1 | 0 | 0 | 0 | 100 | 100 | - | 100 | - | - | - | |
| 19651LLN | 10 | 0 | 10 | 0 | 0 | 0 | 0 | 100 | - | 100 | - | - | - | - | |
| 80002 | 70 | 2 | 25 | 13 | 1 | 9 | 20 | 54 | 50 | 52 | 100 | 100 | 56 | 25 | |
| MetCRPC | 1135 | 6 | 2 | 4 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | - | - | - | - |
| Nearby Gene | Variant Positions | Data from Literature | Reference | Patient IDs |
|---|---|---|---|---|
| NEAT1 | Chr11:65,190,268-65,213,009 | 13/112 cases, 6/20 mets all with previous ADT. NEAT1 produces a long non-coding RNA that regulates several growth pathways and overexpression is associated with PCa progression | Wedge [8] | 5545 |
| FOXA1 | Promoter, Chr14:37587200-37597201 | 14 coding and 6 non-coding mutations; regulates AR signalling | Wedge | 5684 |
| FOXA1 | Chr14:37886261-37888565, 37903630-37906634, 38035667-38036817, 38053354-38056060, 38056084-38059097, 38127358-38128083 | FOXA1 is a co-factor for AR. These are cis-regulatory elements | Zhou [34] | 5684 |
| AR | Upstream promoter | Tandem duplications, 70–87% mCRPC vs. <2% primary PCa | Viswanathan [11] | Nil |
| AR | ChrX: 66117800-66128800 (66.10–66.20 bin) | I peak, long range enhancer of AR, only 1/54 primary samples (Viswanathan); Copy number gain results in proliferation in low androgen condition and enzalutamide resistance | Takeda [35], Viswanathan | Nil |
| AR | Transcription Factor Binding Sites | Recurrently altered in primary PCa | Morova [33] | 1135, 5545, 5684, 12543, 13179, 19011, 19145, 19260, 80002, 19651 (LP, RP, RLN), A153, PCSD13 |
| MYC | Chr8: 128.14–128.28, 128.47–128.54, 128.54–128.62 | 8q24 risk loci PCa, associated with MYC enhancer activity | Ahmadiyeh [37], Yeager [36] | 19651LP, 12543, A153 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crumbaker, M.; Chan, E.K.F.; Gong, T.; Corcoran, N.; Jaratlerdsiri, W.; Lyons, R.J.; Haynes, A.-M.; Kulidjian, A.A.; Kalsbeek, A.M.F.; Petersen, D.C.; et al. The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis. Cancers 2020, 12, 1178. https://doi.org/10.3390/cancers12051178
Crumbaker M, Chan EKF, Gong T, Corcoran N, Jaratlerdsiri W, Lyons RJ, Haynes A-M, Kulidjian AA, Kalsbeek AMF, Petersen DC, et al. The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis. Cancers. 2020; 12(5):1178. https://doi.org/10.3390/cancers12051178
Chicago/Turabian StyleCrumbaker, Megan, Eva K. F. Chan, Tingting Gong, Niall Corcoran, Weerachai Jaratlerdsiri, Ruth J. Lyons, Anne-Maree Haynes, Anna A. Kulidjian, Anton M. F. Kalsbeek, Desiree C. Petersen, and et al. 2020. "The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis" Cancers 12, no. 5: 1178. https://doi.org/10.3390/cancers12051178
APA StyleCrumbaker, M., Chan, E. K. F., Gong, T., Corcoran, N., Jaratlerdsiri, W., Lyons, R. J., Haynes, A. -M., Kulidjian, A. A., Kalsbeek, A. M. F., Petersen, D. C., Stricker, P. D., Jamieson, C. A. M., Croucher, P. I., Hovens, C. M., Joshua, A. M., & Hayes, V. M. (2020). The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis. Cancers, 12(5), 1178. https://doi.org/10.3390/cancers12051178