Dysregulation of Rho GTPases in Human Cancers



Abstract
:1. Introduction
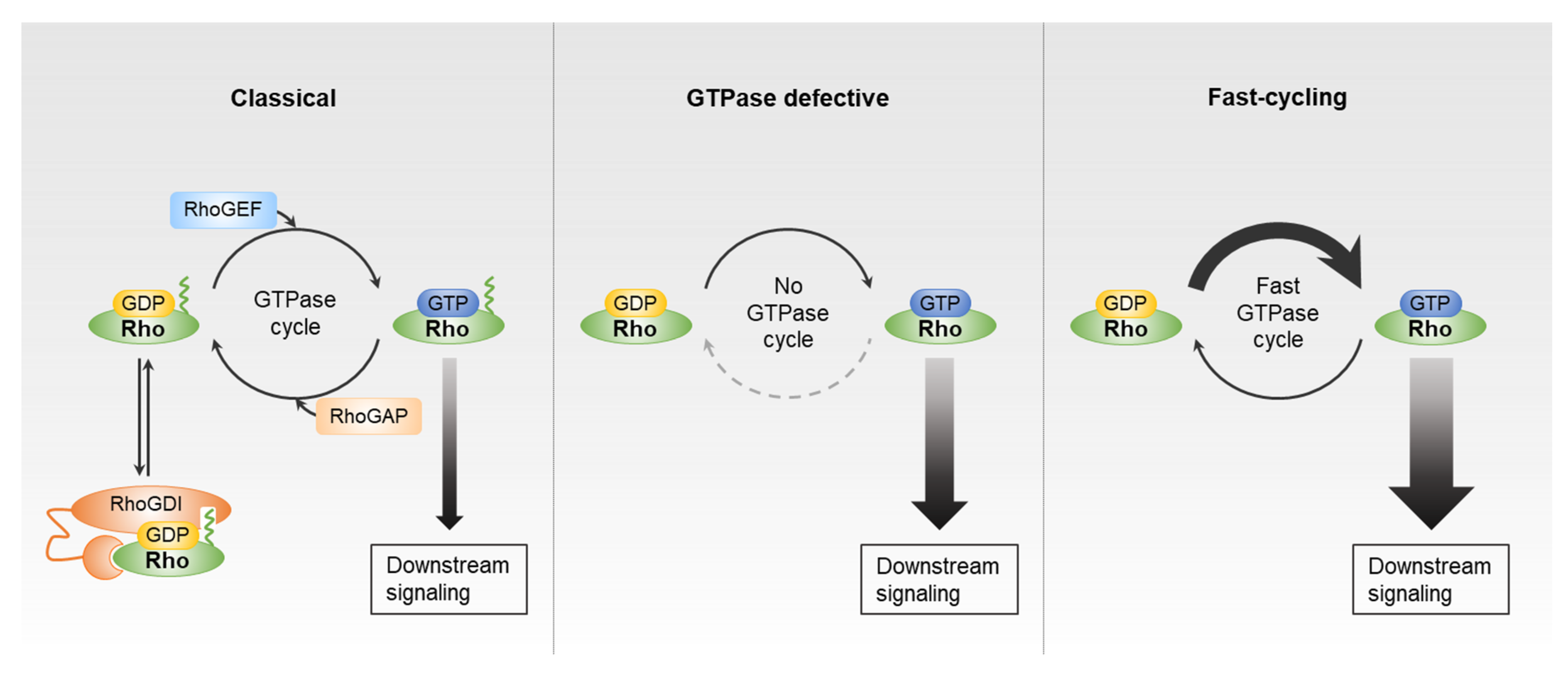
2. Rho GTPases and Their Direct Regulator Partners
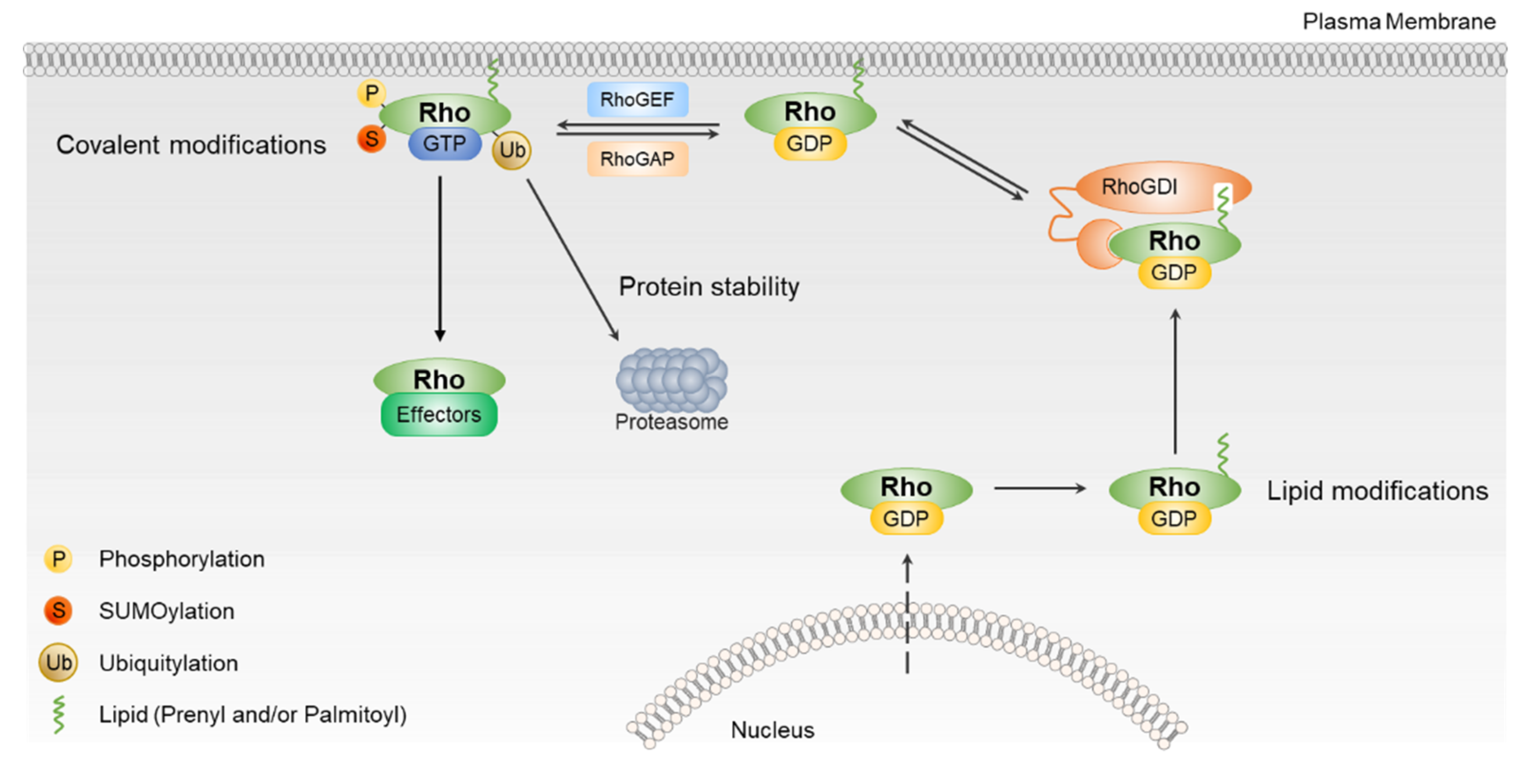
3. Regulation of Rho GTPases by PTMs
3.1. Regulation of Rho GTPases by Lipid Modifications
3.2. Regulation of Rho GTPases by Phosphorylation
3.3. Regulation of Rho GTPases by Ubiquitination and Sumoylation
4. Dysregulation of Rho GTPase Signaling in Human Cancers
4.1. Altered Expression and Activity of Rho GTPases in Human Cancers
4.2. Modulation of Rho GTPase Activity by Regulatory Proteins
4.3. Mutations of Rho GTPases in Human Cancers
5. Therapeutic Targeting of Rho GTPase Signaling
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell. Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.Y.; Zheng, Y. Rho GTPase-activation proteins in cell regulation. Trends Cell Biol. 2003, 13, 13–22. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.; Baek, K.E.; Kim, B.; Lee, H.G. Regulation of Rho GTPases by RhoGDIs in Human Cancers. Cells 2019, 8, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S.; Hall, A. RhoGTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in cancer: Friend or foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- Dransart, E.; Olofsson, B.; Cherfils, J. RhoGDIs revisited: Novel roles in Rho regulation. Traffic 2005, 6, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Golding, A.E.; Visco, I.; Bieling, P.; Bement, W.M. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. eLife 2019, 8, e50471. [Google Scholar] [CrossRef]
- Boulter, E.; Garcia-Mata, R.; Guilluy, C.; Dubash, A.; Rossi, G.; Brennwald, P.J.; Burridge, K. Regulation of RhoGTPase crosstalk, degradation and activity by RhoGDI1. Nat. Cell Biol. 2010, 12, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Baek, K.E.; Yoo, J. RhoGDI2 as a therapeutic target in cancer. Expert Opin. Ther. Targets 2010, 14, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Mouly, L.; Gilhodes, J.; Lemarié, A.; Cohen-Jonathan Moyal, E.; Toulas, C.; Favre, G.; Sordet, O.; Monferran, S. The RND1 Small GTPase: Main Functions and Emerging Role in Oncogenesis. Int. J. Mol. Sci. 2019, 20, 3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Rivero, F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells 2016, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspenström, P.; Ruusala, A.; Pacholsky, D. Taking Rho GTPases to the next level: The cellular functions of atypical Rho GTPases. Exp. Cell. Res. 2007, 313, 3673–3679. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulation and functions of RhoU and RhoV. Small GTPases 2020, 11, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Gad, A.K.; Aspenström, P. Rif proteins take to the RhoD: Rho GTPases at the crossroads of actin dynamics and membrane trafficking. Cell Signal 2010, 22, 183–189. [Google Scholar] [CrossRef]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef]
- Aspenström, P. Activated Rho GTPases in Cancer-The Beginning of a New Paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.; Gesbert, F.; Delespine-Carmagnat, M.; Stancou, R.; Pouchelet, M.; Bertoglio, J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996, 15, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, E.; Sabouri-Ghomi, M.; Pertz, O.; Kim, C.; Gutierrez, E.; Machacek, M.; Groisman, A.; Danuser, G.; Ginsberg, M.H. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat. Cell Biol. 2011, 13, 660–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolli-Derkinderen, M.; Sauzeau, V.; Boyer, L.; Lemichez, E.; Baron, C.; Henrion, D.; Loirand, G.; Pacaud, P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 2005, 96, 1152–1160. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, S.; Chen, S.; Chen, S.; Geng, Q.; Xu, D. c-Met-dependent phosphorylation of RhoA plays a key role in gastric cancer tumorigenesis. J. Pathol. 2019, 249, 126–136. [Google Scholar] [CrossRef]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation of Rac1 T108 by extracellular signal-regulated kinase in response to epidermal growth factor: A novel mechanism to regulate Rac1 function. Mol. Cell Biol. 2013, 33, 4538–4551. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Lemmon, C.; Lietha, D.; Eck, M.; Romer, L. Tyrosine phosphorylation of Rac1: A role in regulation of cell spreading. PLoS ONE 2011, 6, e28587. [Google Scholar] [CrossRef] [Green Version]
- Kwon, T.; Kwon, D.Y.; Chun, J.; Kim, J.H.; Kang, S.S. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J. Biol. Chem. 2000, 275, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.; Wu, W.J.; Wang, J.; Cerione, R.A. Epidermal Growth Factor-dependent Regulation of Cdc42 Is Mediated by the Src Tyrosine Kinase. J. Biol. Chem. 2003, 278, 49293–49300. [Google Scholar] [CrossRef] [Green Version]
- Forget, M.A.; Desrosiers, R.R.; Gingras, D.; Béliveau, R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem. J. 2002, 361, 243–254. [Google Scholar] [CrossRef]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Mialki, R.K.; Dong, S.; Khoo, A.; Mallampalli, R.K.; Zhao, Y.; Zhao, J. A new mechanism of RhoA ubiquitination and degradation: Roles of SCF(FBXL19) E3 ligase and Erk2. Biochim. Biophys. Acta 2013, 1833, 2757–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, T.K.; Dogan, T.; Hocking, J.C.; Scholz, R.P.; Mooz, J.; Anderson, C.L.; Karreman, C.; Meyer zu Heringdorf, D.; Schmidt, G.; Ruonala, M.; et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012, 31, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Mialki, R.K.; Wei, J.; Coon, T.A.; Zou, C.; Chen, B.B.; Mallampalli, R.K.; Zhao, Y. SCF E3 ligase F-box protein complex SCF(FBXL19) regulates cell migration by mediating Rac1 ubiquitination and degradation. FASEB J. 2013, 27, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Lerm, M.; Schmidt, G.; Goehring, U.M.; Schirmer, J.; Aktories, K. Identification of the region of rho involved in substrate recognition by Escherichia coli cytotoxic necrotizing factor 1 (CNF1). J. Biol. Chem. 1999, 274, 28999–29004. [Google Scholar] [CrossRef]
- Yarbrough, M.L.; Li, Y.; Kinch, L.N.; Grishin, N.V.; Ball, H.L.; Orth, K. AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 2009, 323, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell Biol. 2010, 12, 1078–1085. [Google Scholar] [CrossRef]
- Yue, X.; Zhang, C.; Zhao, Y.; Liu, J.; Lin, A.W.; Tan, V.M.; Drake, J.M.; Liu, L.; Boateng, M.N.; Li, J.; et al. Gain-of-function mutant p53 activates small GTPase Rac1 through SUMOylation to promote tumor progression. Genes Dev. 2017, 31, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Katayama, M.; Kawata, M.; Yoshida, Y.; Horiuchi, H.; Yamamoto, T.; Matsuura, Y.; Takai, Y. The posttranslationally modified C-terminal structure of bovine aortic smooth muscle rhoA p21. J. Biol. Chem. 1991, 266, 12639–12645. [Google Scholar] [PubMed]
- Adamson, P.; Marshall, C.J.; Hall, A.; Tilbrook, P.A. Post-translational modifications of p21rho proteins. J. Biol. Chem. 1992, 267, 20033–20038. [Google Scholar]
- Keep, N.H.; Barnes, M.; Barsukov, I.; Badii, R.; Lian, L.Y.; Segal, A.W.; Moody, P.C.; Roberts, G.C. A modulator of rho family G proteins, rhoGDI, binds these G proteins via an immunoglobulin-like domain and a flexible N-terminal arm. Structure 1997, 5, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Gosser, Y.Q.; Nomanbhoy, T.K.; Aghazadeh, B.; Manor, D.; Combs, C.; Cerione, R.A.; Rosen, M.K. C-terminal binding domain of Rho GDP-dissociation inhibitor directs N-terminal inhibitory peptide to GTPases. Nature 1997, 387, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.E.; Deschenes, R. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Berzat, A.C.; Buss, J.E.; Chenette, E.J.; Weinbaum, C.A.; Shutes, A.; Der, C.J.; Minden, A.; Cox, A.D. Transforming activity of the Rho family GTPase, Wrch-1, a Wnt-regulated Cdc42 homolog, is dependent on a novel carboxyl-terminal palmitoylation motif. J. Biol. Chem. 2005, 280, 33055–33065. [Google Scholar] [CrossRef] [Green Version]
- Chenette, E.J.; Mitin, N.Y.; Der, C.J. Multiple sequence elements facilitate Chp Rho GTPase subcellular location, membrane association, and transforming activity. Mol. Biol. Cell 2006, 17, 3108–3121. [Google Scholar] [CrossRef]
- Alan, J.K.; Berzat, A.C.; Dewar, B.J.; Graves, L.M.; Cox, A.D. Regulation of the Rho family small GTPase Wrch-1/RhoU by C-terminal tyrosine phosphorylation requires Src. Mol. Cell Biol. 2010, 30, 4324–4338. [Google Scholar] [CrossRef] [Green Version]
- Riento, K.; Totty, N.; Villalonga, P.; Garg, R.; Guasch, R.; Ridley, A.J. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005, 24, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Madigan, J.P.; Bodemann, B.O.; Brady, D.C.; Dewar, B.J.; Keller, P.J.; Leitges, M.; Philips, M.R.; Ridley, A.J.; Der, C.J.; Cox, A.D. Regulation of Rnd3 localization and function by protein kinase C alpha-mediated phosphorylation. Biochem. J. 2009, 424, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Riou, P.; Kjær, S.; Garg, R.; Purkiss, A.; George, R.; Cain, R.J.; Bineva, G.; Reymond, N.; McColl, B.; Thompson, A.J.; et al. 14-3-3 proteins interact with a hybrid prenyl-phosphorylation motif to inhibit G proteins. Cell 2013, 153, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Vega, M.; Burrows, J.F.; Johnston, J.A. Ubiquitination: Added complexity in Ras and Rho family GTPase function. Small GTPases 2011, 2, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.F. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 2018, 9, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Nethe, M.; Hordijk, P.L. The role of ubiquitylation and degradation in RhoGTPase signalling. J. Cell Sci. 2010, 123, 4011–4018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.R.; Zhang, Y.; Ozdamar, B.; Ogunjimi, A.A.; Alexandrova, E.; Thomsen, G.H.; Wrana, J.L. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003, 302, 1775–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthold, J.; Schenková, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenström, P.; Rivero, F. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes—evidence for an autoregulatory mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef] [Green Version]
- Senadheera, D.; Haataja, L.; Groffen, J.; Heisterkamp, N. The small GTPase Rac interacts with ubiquitination complex proteins Cullin-1 and CDC23. Int. J. Mol. Med. 2001, 8, 127–133. [Google Scholar] [CrossRef]
- Kovacic, H.N.; Irani, K.; Goldschmidt-Clermont, P.J. Redox regulation of human Rac1 stability by the proteasome in human aortic endothelial cells. J. Biol. Chem. 2001, 276, 45856–45861. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, A.; Ping, Q.; Carpenter, C.L. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004, 18, 856–861. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer. Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Huang, C. E3 ubiquitin ligases in regulating stress fiber, lamellipodium, and focal adhesion dynamics. Cell Adh Migr. 2014, 8, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez del Pulgar, T.; Benitah, S.A.; Valerón, P.F.; Espina, C.; Lacal, J.C. Rho GTPase expression in tumourigenesis: Evidence for a significant link. Bioessays 2005, 27, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C.; et al. Anti-RhoA and Anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef]
- Liu, N.; Bi, F.; Pan, Y.; Sun, L.; Xue, Y.; Shi, Y.; Yao, X.; Zheng, Y.; Fan, D. Reversal of the malignant phenotype of gastric cancer cells by inhibition of RhoA expression and activity. Clin. Cancer Res. 2004, 10, 6239–6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Tang, Q.; Xu, F.; Xue, Y.; Zhen, Z.; Deng, Y.; Liu, M.; Chen, J.; Liu, S.; Qiu, M.; et al. RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol. Cancer Res. 2009, 7, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, J.A.; Gilkes, D.M. RhoB: Team Oncogene or Team Tumor Suppressor? Genes 2018, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhu, Y.; Zhang, G.; Liu, N.; Sun, L.; Liu, M.; Qiu, M.; Luo, D.; Tang, Q.; Liao, Z.; et al. A distinct role of RhoB in gastric cancer suppression. Int. J. Cancer 2011, 128, 1057–1068. [Google Scholar] [CrossRef]
- Mazieres, J.; Antonia, T.; Daste, G.; Muro-Cacho, C.; Berchery, D.; Tillement, V.; Pradines, A.; Sebti, S.; Favre, G. Loss of RhoB expression in human lung cancer progression. Clin. Cancer Res. 2004, 10, 2742–2750. [Google Scholar] [CrossRef] [Green Version]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Feng, X.; Shi, M.; Cai, Q.; Yu, Y.; Zhu, Z.; Zhang, J. Rac1 is correlated with aggressiveness and a potential therapeutic target for gastric cancer. Int. J. Oncol. 2015, 46, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.; Cho, S.J.; Chang, K.K.; Park, D.J.; Ryeom, S.W.; Yoon, S.S. Role of Rac1 Pathway in Epithelial-to-Mesenchymal Transition and Cancer Stem-like Cell Phenotypes in Gastric Adenocarcinoma. Mol. Cancer Res. 2017, 15, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Kim, J.T.; Lee, S.J.; Hwang, Y.S.; Park, S.Y.; Kim, B.Y.; Yoo, J.; Hong, K.S.; Min, J.K.; Lee, C.H.; et al. Protein phosphatase 1B dephosphorylates Rho guanine nucleotide dissociation inhibitor 1 and suppresses cancer cell migration and invasion. Cancer Lett. 2018, 417, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Haeusler, L.-C.; Blumenstein, L.; Herbrand, U.; Dvorsky, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 279, 4743–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [Green Version]
- Gómez Del Pulgar, T.; Valdés-Mora, F.; Bandrés, E.; Pérez-Palacios, R.; Espina, C.; Cejas, P.; García-Cabezas, M.A.; Nistal, M.; Casado, E.; González-Barón, M.; et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 2008, 33, 185–193. [Google Scholar] [CrossRef]
- Van Henge, J.; D’Hooge, P.; Hooghe, B.; Wu, X.; Libbrecht, L.; De Vos, R.; Quondamatteo, F.; Klempt, M.; Brakebusch, C.; van Roy, F. Continuous cell injury promotes hepatic tumorigenesis in cdc42-deficient mouse liver. Gastroenterology 2008, 134, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Croisé, P.; Houy, S.; Gand, M.; Lanoix, J.; Calco, V.; Tóth, P.; Brunaud, L.; Lomazzi, S.; Paramithiotis, E.; Chelsky, D.; et al. Cdc42 and Rac1 activity is reduced in human pheochromocytoma and correlates with FARP1 and ARHGEF1 expression. Endocr. Relat. Cancer 2016, 23, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beder, L.B.; Gunduz, M.; Ouchida, M.; Gunduz, E.; Sakai, A.; Fukushima, K.; Nagatsuka, H.; Ito, S.; Honjo, N.; Nishizaki, K.; et al. Identification of a candidate tumor suppressor gene RHOBTB1 located at a novel allelic loss region 10q21 in head and neck cancer. J. Cancer Res. Clin. Oncol. 2006, 132, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Wang, C.; Fu, F.; Chen, Q. RhoBTB2 gene in breast cancer is silenced by promoter methylation. Int. J. Mol. Med. 2014, 33, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grise, F.; Sena, S.; Bidaud-Meynard, A.; Baud, J.; Hiriart, J.B.; Makki, K.; Dugot-Senant, N.; Staedel, C.; Bioulac-Sage, P.; Zucman-Rossi, J.; et al. Rnd3/RhoE Is down-regulated in hepatocellular carcinoma and controls cellular invasion. Hepatology 2012, 55, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, K.; Gu, Y.; Feng, B.; Ren, G.; Zhang, L.; Wang, Y.; Nie, Y.; Fan, D. Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem. Biophys. Res. Commun. 2011, 415, 348–354. [Google Scholar] [CrossRef]
- Li, K.; Lu, Y.; Liang, J.; Luo, G.; Ren, G.; Wang, X.; Fan, D. RhoE enhances multidrug resistance of gastric cancer cells by suppressing Bax. Biochem. Biophys. Res. Commun. 2009, 379, 212–216. [Google Scholar] [CrossRef]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Hirata, D.; Yamabuki, T.; Miki, D.; Ito, T.; Tsuchiya, E.; Fujita, M.; Hosokawa, M.; Chayama, K.; Nakamura, Y.; Daigo, Y. Involvement of epithelial cell transforming sequence-2 oncoantigen in lung and esophageal cancer progression. Clin. Cancer Res. 2009, 15, 256–266. [Google Scholar] [CrossRef] [Green Version]
- Huff, L.P.; Decristo, M.J.; Trembath, D.; Kuan, P.F.; Yim, M.; Liu, J.; Cook, D.R.; Miller, C.R.; Der, C.J.; Cox, A.D. The Role of Ect2 Nuclear RhoGEF Activity in Ovarian Cancer Cell Transformation. Genes Cancer 2013, 4, 460–475. [Google Scholar] [CrossRef]
- Chen, J.; Xia, H.; Zhang, X.; Karthik, S.; Pratap, S.V.; Ooi, L.L.; Hong, W.; Hui, K.M. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 1287–1295. [Google Scholar] [CrossRef]
- Boissier, P.; Huynh-Do, U. The guanine nucleotide exchange factor Tiam1: A Janus-faced molecule in cellular signaling. Cell Signal. 2014, 26, 483–491. [Google Scholar] [CrossRef]
- Minard, M.E.; Kim, L.S.; Price, J.E.; Gallick, G.E. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res. Treat. 2004, 84, 21–32. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Zhu, H.; Shao, L.; Chen, Y.W. Ankyrin-Tiam1 interaction promotes Rac1 signaling and metastatic breast tumor cell invasion and migration. J. Cell Biol. 2000, 150, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Krasnitz, A.; Lucito, R.; Sordella, R.; Vanaelst, L.; Cordon-Cardo, C.; Singer, S.; Kuehnel, F.; Wigler, M.; Powers, S.; et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008, 22, 1439–1444. [Google Scholar] [CrossRef] [Green Version]
- Seng, T.J.; Low, J.S.; Li, H.; Cui, Y.; Goh, H.K.; Wong, M.L.; Srivastava, G.; Sidransky, D.; Califano, J.; Steenbergen, R.D. The major 8p22 tumor suppressor DLC1 is frequently silenced by methylation in both endemic and sporadic nasopharyngeal, esophageal, and cervical carcinomas, and inhibits tumor cell colony formation. Oncogene 2007, 26, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, G. A tumor suppressor DLC1: The functions and signal pathways. J. Cell Physiol. 2020, 235, 4999–5007. [Google Scholar] [CrossRef]
- Wolf, R.M.; Draghi, N.; Liang, X.; Dai, C.; Uhrbom, L.; Eklöf, C.; Westermark, B.; Holland, E.C.; Resh, M.D. p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: A putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev. 2003, 17, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; Guo, J.; Chen, L.; Yang, W.; Qu, X.; Cheng, Z. ARHGAP10, downregulated in ovarian cancer, suppresses tumorigenicity of ovarian cancer cells. Cell Death Dis. 2016, 7, e2157. [Google Scholar] [CrossRef]
- Teng, J.P.; Yang, Z.Y.; Zhu, Y.M.; Ni, D.; Zhu, Z.J.; Li, X.Q. The roles of ARHGAP10 in the proliferation, migration and invasion of lung cancer cells. Oncol. Lett. 2017, 14, 4613–4618. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Chen, X.; Jin, Y.; Lu, J.; Cai, Y.; Wei, O.; Zhao, J.; Zhang, W.; Wen, X.; Wang, Y.; et al. Expression of ARHGAP10 correlates with prognosis of prostate cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 3839–3846. [Google Scholar]
- Li, Y.; Zeng, B.; Li, Y.; Zhang, C.; Ren, G. Downregulated expression of ARHGAP10 correlates with advanced stage and high Ki-67 index in breast cancer. PeerJ 2019, 7, e7431. [Google Scholar] [CrossRef]
- Dong, G.; Wang, B.; An, Y.; Li, J.; Wang, X.; Jia, J.; Yang, Q. SIRT1 suppresses the migration and invasion of gastric cancer by regulating ARHGAP5 expression. Cell Death Dis. 2018, 9, 977. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Castellví-Bel, S.; Chang, L.M.; Bessa, X.; Nakagawa, H.; Harada, H.; Sung, R.K.; Piqué, J.M.; Castells, A.; Rustgi, A.K. ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/CDC42GAP/p50RHOGAP: Mutation and expression analyses in colorectal and breast cancers. Gene 2004, 336, 59–71. [Google Scholar] [CrossRef]
- Hu, Q.; Lin, X.; Ding, L.; Zeng, Y.; Pang, D.; Ouyang, N.; Xiang, Y.; Yao, H. ARHGAP42 promotes cell migration and invasion involving PI3K/Akt signaling pathway in nasopharyngeal carcinoma. Cancer Med. 2018, 7, 3862–3874. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Liao, Q.; An, H.; Li, W.; Jin, X.; Cui, S.; Zhao, L. Overexpression of RhoGDI, a novel predictor of distant metastasis, promotes cell proliferation and migration in hepatocellular carcinoma. FEBS Lett. 2014, 588, 503–508. [Google Scholar] [CrossRef]
- Song, Q.; Xu, Y.; Yang, C.; Chen, Z.; Jia, C.; Chen, J.; Zhang, Y.; Lai, P.; Fan, X.; Zhou, X.; et al. miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res. 2014, 74, 3031–3042. [Google Scholar] [CrossRef] [Green Version]
- Harding, M.A.; Theodorescu, D. RhoGDI signaling provides targets for cancer therapy. Eur. J. Cancer 2010, 46, 1252–1259. [Google Scholar] [CrossRef]
- Jiang, W.G.; Watkins, G.; Lane, J.; Cunnick, G.H.; Douglas-Jones, A.; Mokbel, K.; Mansel, R.E. Prognostic value of rho GTPases and rho guanine nucleotide dissociation inhibitors in human breast cancers. Clin. Cancer Res. 2003, 9, 6432–6440. [Google Scholar]
- Gildea, J.J.; Seraj, M.J.; Oxford, G.; Harding, M.A.; Hampton, G.M.; Moskaluk, C.A.; Frierson, H.F.; Conaway, M.R.; Theodorescu, D. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002, 62, 6418–6423. [Google Scholar]
- Cho, H.J.; Baek, K.E.; Park, S.M.; Kim, I.K.; Choi, Y.L.; Cho, H.J.; Nam, I.K.; Hwang, E.M.; Park, J.Y.; Han, J.Y.; et al. RhoGDI2 expression is associated with tumor growth and malignant progression of gastric cancer. Clin. Cancer Res. 2009, 15, 2612–2619. [Google Scholar] [CrossRef] [Green Version]
- Tapper, J.; Kettunen, E.; El-Rifai, W.; Seppälä, M.; Andersson, L.C.; Knuutila, S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet. Cytogenet. 2001, 128, 1–6. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [Green Version]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [Green Version]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.-E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef]
- Stevers, M.; Rabban, J.T.; Garg, K.; Van Ziffl, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Zaloudek, C.; Solomon, D.A. Well-differentiated papillary mesothelioma of the peritoneum is genetically defined by mutually exclusive mutations in TRAF7 and CDC42. Mod. Pathol. 2019, 32, 88–99. [Google Scholar] [CrossRef]
- Mazieres, J.; Pradines, A.; Favre, G. Perspectives on farnesyl transferase inhibitors in cancer therapy. Cancer Lett. 2004, 206, 159–167. [Google Scholar] [CrossRef]
- Chan, K.K.; Oza, A.M.; Siu, L.L. The statins as anticancer agents. Clin. Cancer Res. 2003, 9, 10–19. [Google Scholar]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological actions of statins: A critical appraisal in the management of cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; Chiorean, E.G.; Sebti, S.M.; O’Dwyer, P.J. A Phase I Study of GGTI-2418 (Geranylgeranyl Transferase I Inhibitor) in Patients with Advanced Solid Tumors. Target Oncol. 2019, 14, 613–618. [Google Scholar] [CrossRef]
- Haluska, P.; Dy, G.K.; Adjei, A.A. Farnesyl transferase inhibitors as anticancer agents. Eur. J. Cancer 2002, 38, 1685–1700. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Evelyn, C.R.; Ferng, T.; Rojas, R.J.; Larsen, M.J.; Sondek, J.; Neubig, R.R. High-throughput screening for small-molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J. Biomol. Screen. 2009, 14, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [Green Version]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamori, R.; Yu, S.; Zhang, X.; Hoffman, A.; Sun, J.; Das, S.; Vedula, P.; Li, G.; Fu, J.; Walker, F.; et al. CDC42 inhibition suppresses progression of incipient intestinal tumors. Cancer Res. 2014, 74, 5480–5492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesland, A.; Zhao, Y.; Chen, Y.H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, B.J.; Zhao, Y.; Zhou, H.; Huo, S.; Chen, Y.H.; Lu, Q. Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol. Ther. 2019, 20, 740–749. [Google Scholar] [CrossRef]
- Kale, V.P.; Hengst, J.A.; Desai, D.H.; Amin, S.G.; Yun, J.K. The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett. 2015, 361, 185–196. [Google Scholar] [CrossRef]
- Rath, N.; Olson, M.F. Rho-associated kinases in tumorigenesis: Re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012, 13, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef]
- Nagumo, H.; Sasaki, Y.; Ono, Y.; Okamoto, H.; Seto, M.; Takuwa, Y. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am. J. Physiol. Cell Physiol. 2000, 278, C57–C65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routhier, A.; Astuccio, M.; Lahey, D.; Monfredo, N.; Johnson, A.; Callahan, W.; Partington, A.; Fellows, K.; Ouellette, L.; Zhidro, S.; et al. Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol. Rep. 2010, 23, 861–867. [Google Scholar] [PubMed]
- Ying, H.; Biroc, S.L.; Li, W.W.; Alicke, B.; Xuan, J.A.; Pagila, R.; Ohashi, Y.; Okada, T.; Kamata, Y.; Dinter, H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006, 5, 2158–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.A.; Forinash, K.D.; Pireddu, R.; Sun, Y.; Sun, N.; Martin, M.P.; Schönbrunn, E.; Lawrence, N.J.; Sebti, S.M. RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res. 2012, 72, 5025–5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Hayashi, K.; Egi, Y.; Katayama, K.; Amano, Y.; Uehata, M.; Ohtsuki, M.; Fujii, A.; Oshita, K.; Kataoka, H.; et al. Effect of Wf-536, a novel ROCK inhibitor, against metastasis of B16 melanoma. Cancer Chemother. Pharmacol. 2003, 52, 319–324. [Google Scholar] [CrossRef]
- Yap, T.A.; Walton, M.I.; Grimshaw, K.M.; Te Poele, R.H.; Eve, P.D.; Valenti, M.R.; de Haven Brandon, A.K.; Martins, V.; Zetterlund, A.; Heaton, S.P.; et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin. Cancer Res. 2012, 18, 3912–3923. [Google Scholar] [CrossRef] [Green Version]
- Licciulli, S.; Maksimoska, J.; Zhou, C.; Troutman, S.; Kota, S.; Liu, Q.; Duron, S.; Campbell, D.; Chernoff, J.; Field, J.; et al. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated Schwannomas. J. Biol. Chem. 2013, 288, 29105–29114. [Google Scholar] [CrossRef] [Green Version]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.L.; Lam, I.P.; Wong, T.Y.; Lai, W.L.; Liu, H.F.; Yeung, L.L.; Ching, Y.P. IPA-3 inhibits the growth of liver cancer cells by suppressing PAK1 and NF-κB activation. PLoS ONE 2013, 8, e68843. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gratzke, C.; Tamalunas, A.; Wiemer, N.; Ciotkowska, A.; Rutz, B.; Waidelich, R.; Strittmatter, F.; Liu, C.; Stief, C.G.; et al. P21-Activated Kinase Inhibitors FRAX486 and IPA3: Inhibition of Prostate Stromal Cell Growth and Effects on Smooth Muscle Contraction in the Human Prostate. PLoS ONE 2016, 11, e0153312. [Google Scholar] [CrossRef]
- Kaneko, M.; Saito, Y.; Saito, H.; Matsumoto, T.; Matsuda, Y.; Vaught, J.L.; Dionne, C.A.; Angeles, T.S.; Glicksman, M.A.; Neff, N.T.; et al. Neurotrophic 3,9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J. Med. Chem. 1997, 40, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Porchia, L.M.; Guerra, M.; Wang, Y.C.; Zhang, Y.; Espinosa, A.V.; Shinohara, M.; Kulp, S.K.; Kirschner, L.S.; Saji, M.; Chen, C.S.; et al. 2-amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phenyl} acetamide (OSU-03012), a celecoxib derivative, directly targets p21-activated kinase. Mol. Pharmacol. 2007, 72, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| PTMs | Rho GTPase | Regulator | Target Site | Effects | Refs |
|---|---|---|---|---|---|
| Phosphorylation | RhoA | PKA | S188 | Increasing interaction with RhoGDI | [21,22] |
| Decreases binding to ROCK effector | |||||
| Protection of RhoA from degradation | |||||
| PKG | S188 | Translocation to the cytosol | [23] | ||
| Protection of RhoA from degradation | |||||
| PKC | T127 and S188 | Translocation to the plasma membrane | [24] | ||
| Protection of RhoA from degradation | |||||
| c-Met | Tyr42 | Proteasomal degradation | [25] | ||
| Rac1 | ERK | T108 | RAC1 for translocation to the nucleus | [26] | |
| FAK | Y64 | Inhibit RAC1 activity | [27] | ||
| SRC | Y64 | Inhibit RAC1 activity | [27] | ||
| AKT | S71 | Inhibits RAC1 activity | [28] | ||
| Cdc42 | SRC | Y64 | Increasing interaction with RhoGDI | [29] | |
| PKA | S185 | Increasing interaction with RhoGDI | [30] | ||
| Ubiquitination | RhoA | SMURF1 | K6,K7 and K51 | Proteasomal degradation | [31] |
| SCF | K135 | Proteasomal degradation in a ERK-dependent manner | [32] | ||
| CUL3 | ND | Proteasomal degradation of GDP-bound inactive RhoA | [33] | ||
| Rac1 | XIAP and clAP1 | K147 | Proteasomal degradation | [34] | |
| HACE1 | K147 | Targets GTP-bound, active RAC1 for degradation | [35] | ||
| SCF | K166 | Proteasomal degradation in a AKT-dependent manner | [36] | ||
| Transglutamination | RhoA | CNF1 | E63 | Constitutive activation | [37] |
| Rac1 | CNF1 | E61 | Constitutive activation | [37] | |
| Cdc42 | CNF1 | E61 | Constitutive activation | [37] | |
| AMPylation | RhoA | HYPE | T37 | Suppress effector binding | [38] |
| Rac1 | HYPE | T35 | Suppress effector binding | [38] | |
| Cdc42 | HYPE | T35 | Suppress effector binding | [38] | |
| SUMOylation | Rac1 | PIAS3 | K183,K184,K186 and K188 | Increased GTP binding and RAC1 activation | [39,40] |
| Drug Name | Target Protein | Mechanism | Refs |
|---|---|---|---|
| Rhosin | RhoA | Inhibit GEF biding | [129] |
| Y16 | LARG | Inhibit RhoA binding | [130] |
| NSC23766 | Rac1 | Inhibit GEF binding | [132] |
| EHop-016 | Rac1 | Derivative of NSC23766 | [133] |
| CASIN | Cdc42 | Inhibit GEF binding | [134,135] |
| ZCL278 | Cdc42 | Inhibit GEF binding | [136,137] |
| Y-27632 | ROCK | Compete with ATP | [140] |
| Wf-536 | ROCK | Derivative of Y-27632 | [144] |
| Fasudil | ROCK | Compete with ATP | [141] |
| RKI-1447 | ROCK | Compete with ATP | [144] |
| AT13148 | ROCK | Compete with ATP | [146] |
| K252 | PAK | Compete with ATP | [151] |
| OSU-03012 | PAK | Compete with ATP | [152] |
| PF-3758309 | PAK | Compete with ATP | [153] |
| FRAX597 | PAK | Compete with ATP | [147] |
| IPA-3 | PAK | Promote auto-inhibited conformation | [148] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.; Yoon, S.R.; Lim, J.; Cho, H.J.; Lee, H.G. Dysregulation of Rho GTPases in Human Cancers. Cancers 2020, 12, 1179. https://doi.org/10.3390/cancers12051179
Jung H, Yoon SR, Lim J, Cho HJ, Lee HG. Dysregulation of Rho GTPases in Human Cancers. Cancers. 2020; 12(5):1179. https://doi.org/10.3390/cancers12051179
Chicago/Turabian StyleJung, Haiyoung, Suk Ran Yoon, Jeewon Lim, Hee Jun Cho, and Hee Gu Lee. 2020. "Dysregulation of Rho GTPases in Human Cancers" Cancers 12, no. 5: 1179. https://doi.org/10.3390/cancers12051179
APA StyleJung, H., Yoon, S. R., Lim, J., Cho, H. J., & Lee, H. G. (2020). Dysregulation of Rho GTPases in Human Cancers. Cancers, 12(5), 1179. https://doi.org/10.3390/cancers12051179