Hypoxic Roadmap of Glioblastoma—Learning about Directions and Distances in the Brain Tumor Environment




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
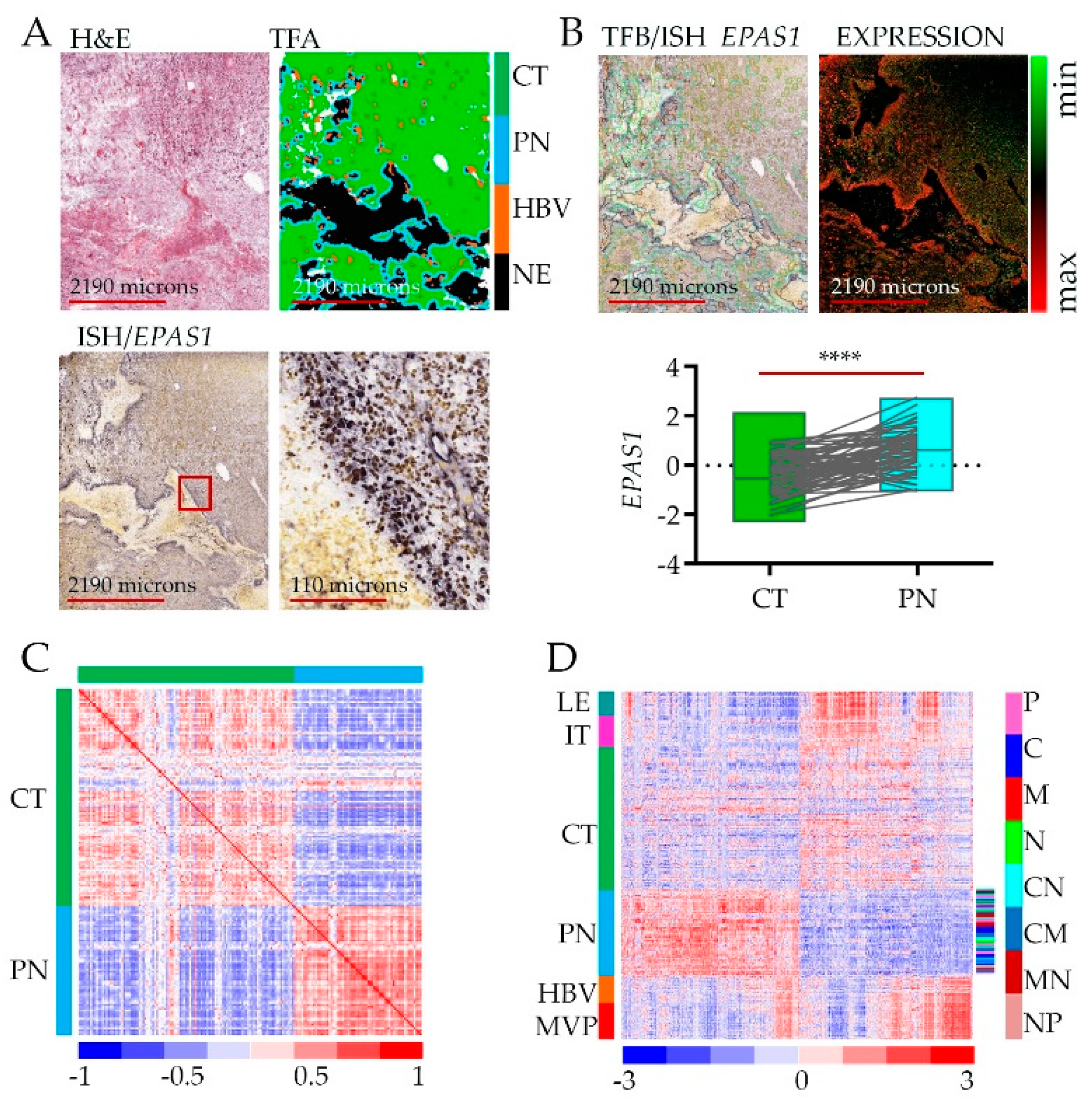
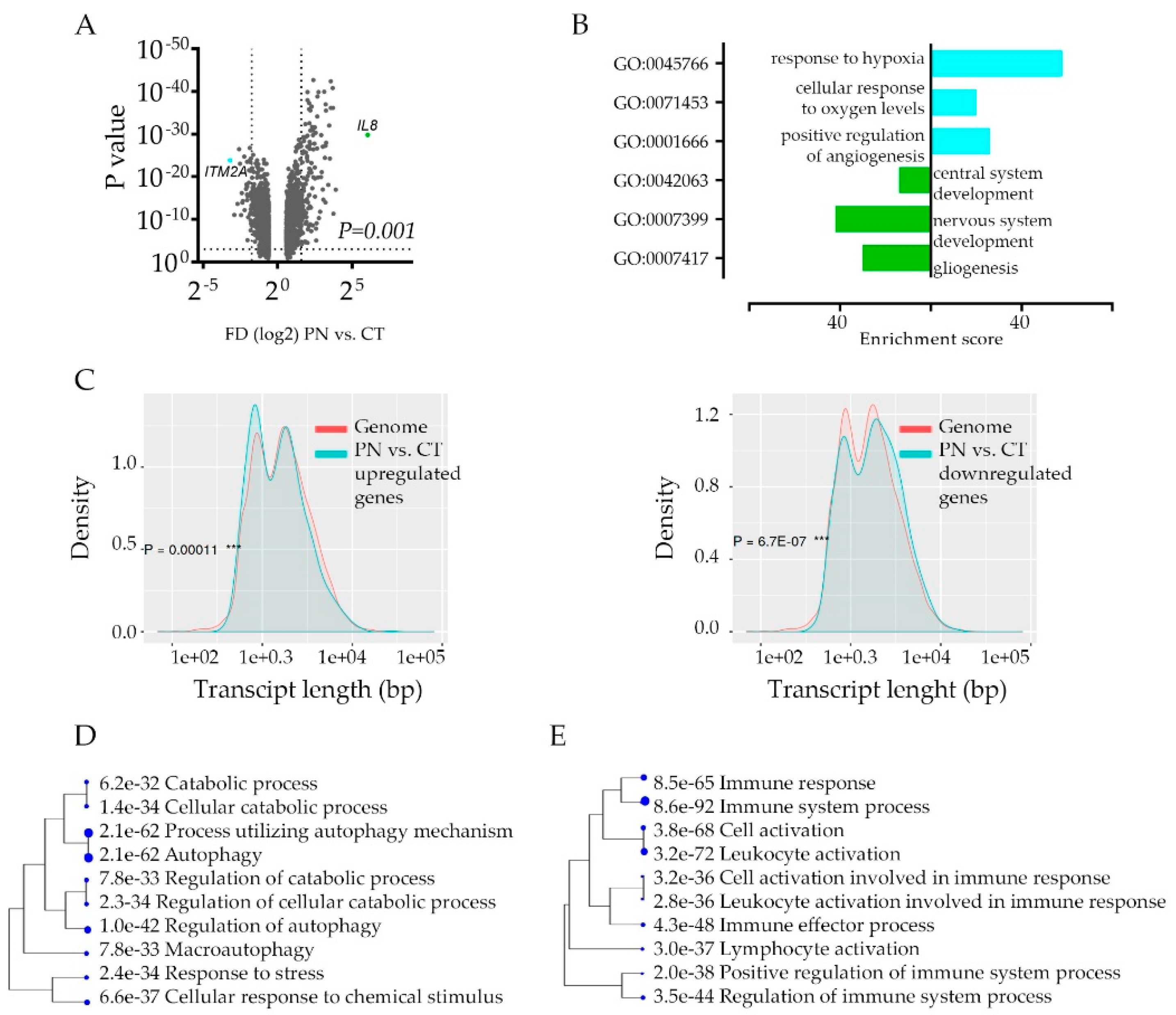
2.1. Hypoxic Zone—Gain and Loss of Transcriptome
2.2. Gene Ontology Analysis of the Hypoxic Zone—Adaptation or Defense
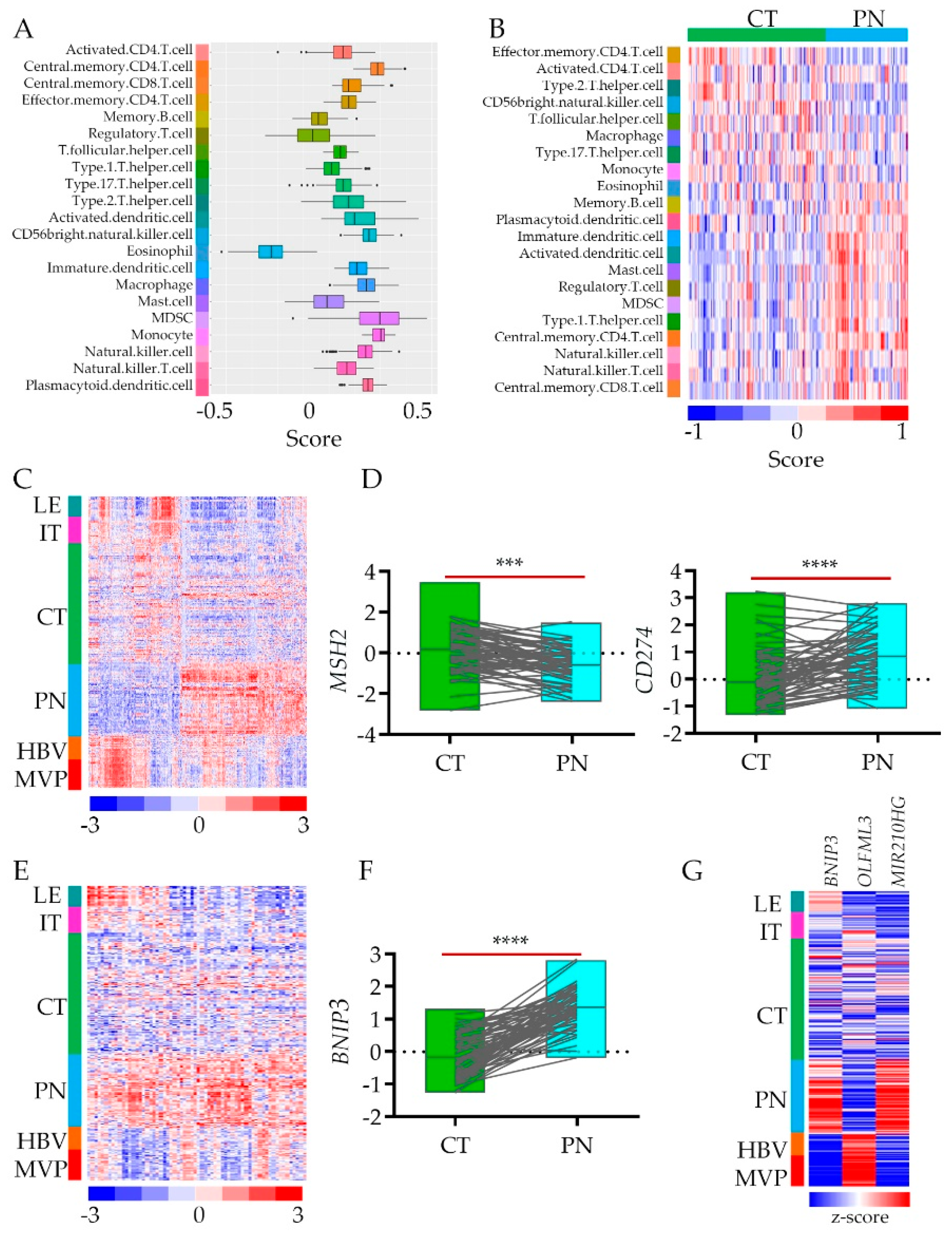
2.3. Tumors’ Hypoxic Zones Require a Systemic Approach
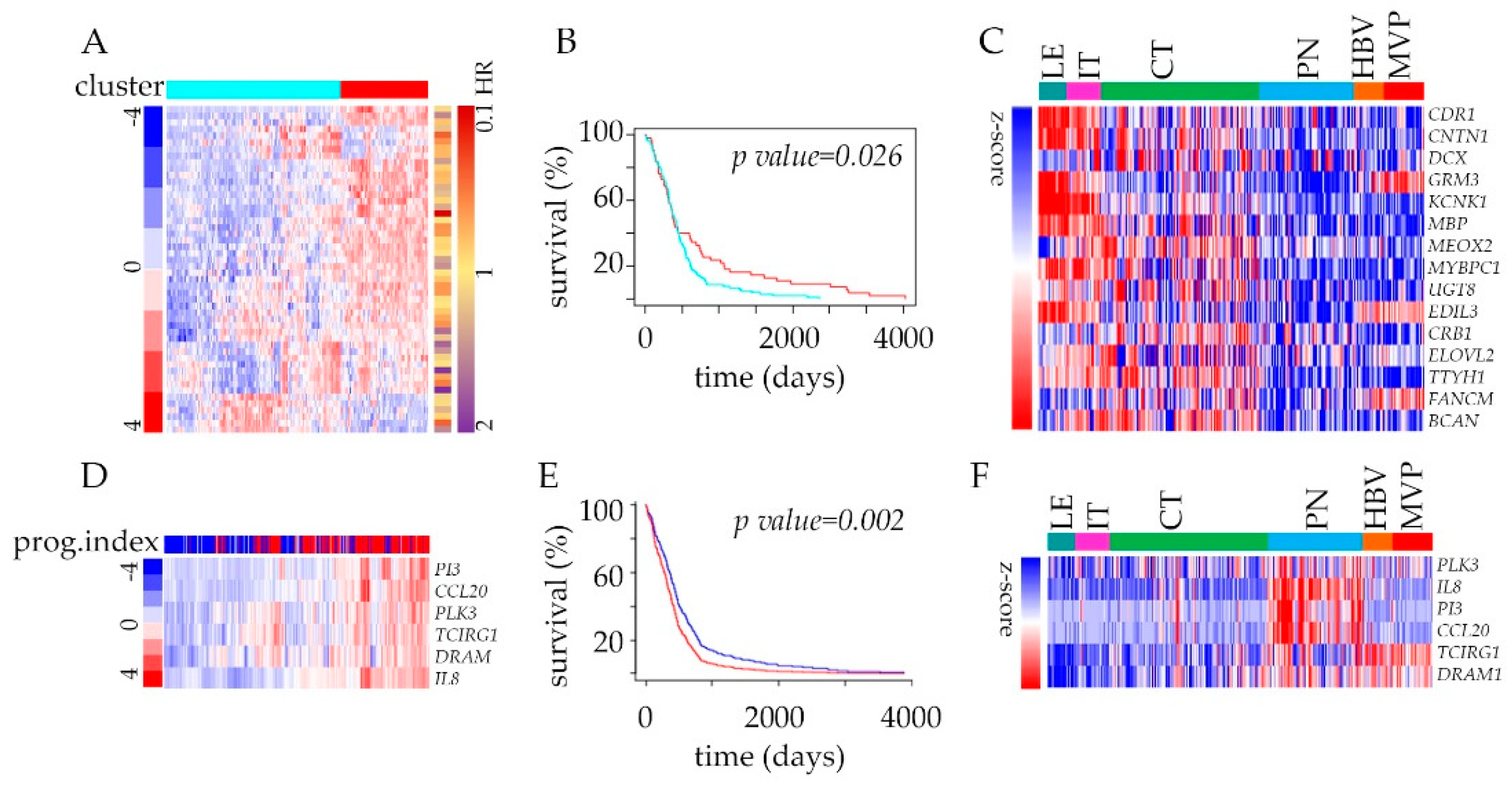
2.4. The Impact of Hypoxia on the Outcome of Glioblastoma Patients
3. Discussion
4. Materials and Methods
Databases and Data Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Wucherpfennig, K.; Chiocca, E.A. Immunotherapy for glioblastoma: On the sidelines or in the game? Discov. Med. 2017, 24, 201–208. [Google Scholar]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Babikir, H.; Muller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef] [Green Version]
- Rooj, A.K.; Ricklefs, F.; Mineo, M.; Nakano, I.; Chiocca, E.A.; Bronisz, A.; Godlewski, J. MicroRNA-Mediated Dynamic Bidirectional Shift between the Subclasses of Glioblastoma Stem-like Cells. Cell Rep. 2017, 19, 2026–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godlewski, J.; Ferrer-Luna, R.; Rooj, A.K.; Mineo, M.; Ricklefs, F.; Takeda, Y.S.; Nowicki, M.O.; Salinska, E.; Nakano, I.; Lee, H.; et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep. 2017, 8, 1497–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, N.; Jaffee, E.M. Immune cells track hard-to-target brain tumours. Nature 2019, 565, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Berchem, G.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy 2012, 8, 704–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 68. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, D.; Ansari, K.; Nowicki, M.O.; Salinska, E.; Bronisz, A.; Godlewski, J. MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Non-Cod. RNA 2019, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Thauland, T.J.; Butte, M.J. Taking T cell priming down a Notch: Signaling through Notch receptors enhances T cell sensitivity to antigen. Immunity 2015, 42, 6–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Chi, H. AMPK helps T cells survive nutrient starvation. Immunity 2015, 42, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.; Pham, P.; Dedon, P.C.; Begley, T.J. Lifestyle modifications: Coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol. 2018, 19, 228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Sun, B.Y.; Zhu, Z.H.; Chang, E.T.; To, K.F.; Hwang, J.S.; Jiang, H.; Kam, M.K.; Chen, G.; Cheah, S.L.; et al. Eight-signature classifier for prediction of nasopharyngeal [corrected] carcinoma survival. J. Clin. Oncol. 2011, 29, 4516–4525. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, J.; Han, Z.; Liu, W.; Xi, S.; Huang, L.; Huang, W.; Lin, T.; Zhao, L.; Hu, Y.; et al. Immunomarker Support Vector Machine Classifier for Prediction of Gastric Cancer Survival and Adjuvant Chemotherapeutic Benefit. Clin. Cancer Res. 2018, 24, 5574–5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Nakashima, H.; Chiocca, E.A. Molecular responses to immune checkpoint blockade in glioblastoma. Nat. Med. 2019, 25, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Y.; Xu, K.; Wang, Z.; Fan, X.; Zhang, C.; Li, S.; Qiu, X.; Jiang, T. Relationship between necrotic patterns in glioblastoma and patient survival: Fractal dimension and lacunarity analyses using magnetic resonance imaging. Sci. Rep. 2017, 7, 8302. [Google Scholar] [CrossRef]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Moszynska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Kroliczewski, J.; Dabrowski, M.; et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.H.; Chang, A.; Tan, Z.; Lan, Z.; Zhou, A.; Spring, D.J.; Lang, F.F.; Wang, Y.A.; DePinho, R.A. Circadian regulator CLOCK recruits immune suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tian, C.; Sun, J.; Chen, L.N.; Lv, Y.; Yang, X.D.; Xiao, K.; Wang, J.; Chen, C.; Shi, Q.; et al. Overexpression of PLK3 Mediates the Degradation of Abnormal Prion Proteins Dependent on Chaperone-Mediated Autophagy. Mol. Neurobiol. 2017, 54, 4401–4413. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [Green Version]
- El Hout, M.; Cosialls, E.; Mehrpour, M.; Hamai, A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol. Cancer 2020, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Mineo, M.; Ricklefs, F.; Rooj, A.K.; Lyons, S.M.; Ivanov, P.; Ansari, K.I.; Nakano, I.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 2016, 15, 2500–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Rooj, A.K.; Bronisz, A.; Godlewski, J. The role of octamer binding transcription factors in glioblastoma multiforme. Biochim. Biophys. Acta 2016, 1859, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Bronisz, A.; Chiocca, E.A.; Godlewski, J. Response to energy depletion: miR-451/AMPK loop. Oncotarget 2015, 6, 17851–17852. [Google Scholar] [CrossRef]
- Godlewski, J.; Lenart, J.; Salinska, E. MicroRNA in Brain pathology: Neurodegeneration the Other Side of the Brain Cancer. Noncod. RNA 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bronisz, A.; Salińska, E.; Chiocca, E.A.; Godlewski, J. Hypoxic Roadmap of Glioblastoma—Learning about Directions and Distances in the Brain Tumor Environment. Cancers 2020, 12, 1213. https://doi.org/10.3390/cancers12051213
Bronisz A, Salińska E, Chiocca EA, Godlewski J. Hypoxic Roadmap of Glioblastoma—Learning about Directions and Distances in the Brain Tumor Environment. Cancers. 2020; 12(5):1213. https://doi.org/10.3390/cancers12051213
Chicago/Turabian StyleBronisz, Agnieszka, Elżbieta Salińska, E. Antonio Chiocca, and Jakub Godlewski. 2020. "Hypoxic Roadmap of Glioblastoma—Learning about Directions and Distances in the Brain Tumor Environment" Cancers 12, no. 5: 1213. https://doi.org/10.3390/cancers12051213
APA StyleBronisz, A., Salińska, E., Chiocca, E. A., & Godlewski, J. (2020). Hypoxic Roadmap of Glioblastoma—Learning about Directions and Distances in the Brain Tumor Environment. Cancers, 12(5), 1213. https://doi.org/10.3390/cancers12051213