Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
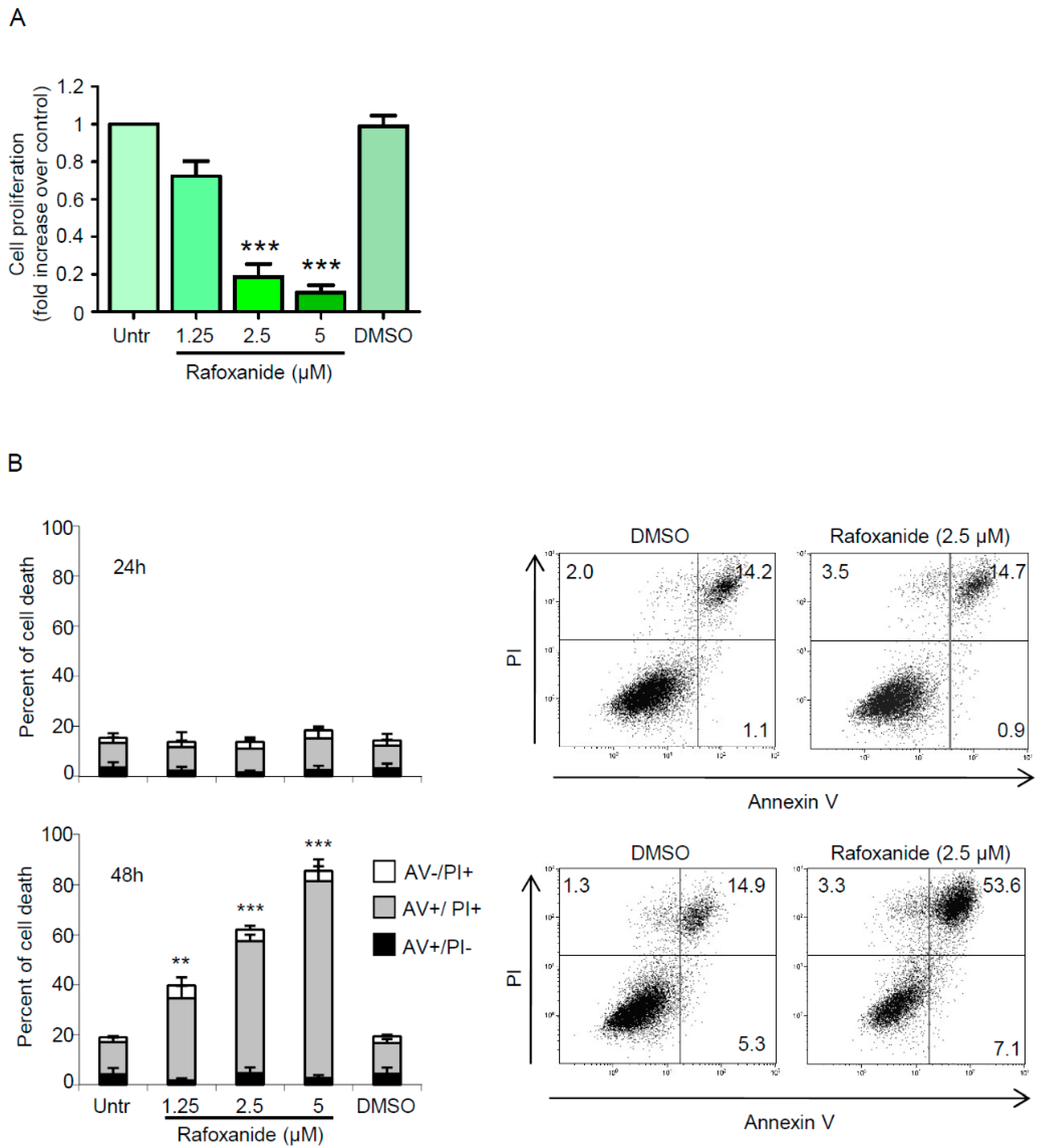
2.1. Treatment of CRC Cells with Rafoxanide Induces Pre-Apoptotic Exposure of CALR on the Cell Surface
2.2. CRC Cells Release ATP and HMGB1 after Rafoxanide Exposure
2.3. Anti-Cancer Effects of Rafoxanide on the Murine Adenocarcinoma Cell Line CT26 Associate with the Induction of ICD Markers
2.4. Rafoxanide Exerts Protective Anti-Cancer Activity in a Vaccination Assay
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blotting
4.3. Assessment of ecto-CALR, ATP and HMGB1
4.4. Assessment of Cell Proliferation and Death
4.5. Tumor Vaccination Assay
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Senovilla, L.; Aranda, F.; Galluzzi, L.; Kroemer, G. Impact of myeloid cells on the efficacy of anticancer chemotherapy. Curr. Opin. Immunol. 2014, 30, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Armour, J.; Corba, J. The anthelmintic activity of rafoxanide against immature Fasciola hepatica in sheep. Vet. Rec. 1970, 87, 213–214. [Google Scholar] [CrossRef]
- Knapp, S.E.; Presidente, P.J. Efficacy of rafoxanide against natural Fasciola hepatica infections in cattle. Am. J. Vet. Res. 1971, 32, 1289–1291. [Google Scholar] [PubMed]
- Swan, G.E. The pharmacology of halogenated salicylanilides and their anthelmintic use in animals. J. S. Afr. Vet. Assoc. 1999, 70, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurdakok, M. Rafanoxide therapy in a child with fascioliasis. Mikrobiyoloji Bul. 1985, 19, 38–40. [Google Scholar]
- Li, Y.; Guo, B.; Xu, Z.; Li, B.; Cai, T.; Zhang, X.; Yu, Y.; Wang, H.; Shi, J.; Zhu, W. Repositioning organohalogen drugs: A case study for identification of potent B-Raf V600E inhibitors via docking and bioassay. Sci. Rep. 2016, 6, 31074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barras, D. BRAF Mutation in Colorectal Cancer: An Update. Biomark. Cancer 2015, 7, 9–12. [Google Scholar] [CrossRef]
- Liu, J.Z.; Hu, Y.L.; Feng, Y.; Guo, Y.B.; Liu, Y.F.; Yang, J.L.; Mao, Q.S.; Xue, W.J. Rafoxanide promotes apoptosis and autophagy of gastric cancer cells by suppressing PI3K /Akt/mTOR pathway. Exp. Cell Res. 2019, 385, 111691. [Google Scholar] [CrossRef]
- Shi, X.; Li, H.; Shi, A.; Yao, H.; Ke, K.; Dong, C.; Zhu, Y.; Qin, Y.; Ding, Y.; He, Y.H.; et al. Discovery of rafoxanide as a dual CDK4/6 inhibitor for the treatment of skin cancer. Oncol. Rep. 2018, 40, 1592–1600. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Xu, Z.; Chang, S.; Li, B.; Yu, D.; Wu, H.; Xie, Y.; Wang, Y.; Xie, B.; Sun, X.; et al. Rafoxanide, an organohalogen drug, triggers apoptosis and cell cycle arrest in multiple myeloma by enhancing DNA damage responses and suppressing the p38 MAPK pathway. Cancer Lett. 2019, 444, 45–59. [Google Scholar] [CrossRef]
- He, W.; Xu, Z.; Song, D.; Zhang, H.; Li, B.; Gao, L.; Zhang, Y.; Feng, Q.; Yu, D.; Hu, L.; et al. Antitumor effects of rafoxanide in diffuse large B cell lymphoma via the PTEN/PI3K/Akt and JNK/c-Jun pathways. Life Sci. 2020, 243, 117249. [Google Scholar] [CrossRef]
- Laudisi, F.; Di Grazia, A.; De Simone, V.; Cherubini, F.; Colantoni, A.; Ortenzi, A.; Franze, E.; Dinallo, V.; Di Fusco, D.; Monteleone, I.; et al. Induction of endoplasmic reticulum stress and inhibition of colon carcinogenesis by the anti-helmintic drug rafoxanide. Cancer Lett. 2019, 462, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Semeraro, M.; Bravo-San Pedro, J.M.; Bloy, N.; Buque, A.; Huang, X.; Zhou, H.; Senovilla, L.; Kroemer, G.; Galluzzi, L. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin. Cancer Biol. 2015, 33, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Iribarren, K.; Michels, J.; Leary, A.; Zitvogel, L.; Cremer, I.; Kroemer, G. Calreticulin expression: Interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology 2016, 5, e1177692. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012, 337, 1678–1684. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Milne, K.; Truong, P.T.; Macpherson, N.; Nelson, B.H.; Watson, P.H. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res. BCR 2011, 13, R126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Juarez, M.; Schcolnik-Cabrera, A.; Duenas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Liu, P.; Zhao, L.; Loos, F.; Iribarren, K.; Lachkar, S.; Zhou, H.; Gomes-da-Silva, L.C.; Chen, G.; Bezu, L.; Boncompain, G.; et al. Identification of pharmacological agents that induce HMGB1 release. Sci. Rep. 2017, 7, 14915. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek-Peric, A.M.; Romano, E.; Agostinis, P. Immunogenic cell death. Int. J. Dev. Biol. 2015, 59, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.; Vacchelli, E.; Aranda, F.; Castoldi, F.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Fucikova, J.; Galon, J.; Spisek, R.; et al. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015, 4, e1008866. [Google Scholar] [CrossRef]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef]
- Emens, L.A. Chemoimmunotherapy. Cancer J. 2010, 16, 295–303. [Google Scholar] [CrossRef]
- Stolfi, C.; Fina, D.; Caruso, R.; Caprioli, F.; Sarra, M.; Fantini, M.C.; Rizzo, A.; Pallone, F.; Monteleone, G. Cyclooxygenase-2-dependent and -independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem. Pharmacol. 2008, 75, 668–676. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Grazia, A.; Laudisi, F.; Di Fusco, D.; Franzè, E.; Ortenzi, A.; Monteleone, I.; Monteleone, G.; Stolfi, C. Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells. Cancers 2020, 12, 1314. https://doi.org/10.3390/cancers12051314
Di Grazia A, Laudisi F, Di Fusco D, Franzè E, Ortenzi A, Monteleone I, Monteleone G, Stolfi C. Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells. Cancers. 2020; 12(5):1314. https://doi.org/10.3390/cancers12051314
Chicago/Turabian StyleDi Grazia, Antonio, Federica Laudisi, Davide Di Fusco, Eleonora Franzè, Angela Ortenzi, Ivan Monteleone, Giovanni Monteleone, and Carmine Stolfi. 2020. "Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells" Cancers 12, no. 5: 1314. https://doi.org/10.3390/cancers12051314
APA StyleDi Grazia, A., Laudisi, F., Di Fusco, D., Franzè, E., Ortenzi, A., Monteleone, I., Monteleone, G., & Stolfi, C. (2020). Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells. Cancers, 12(5), 1314. https://doi.org/10.3390/cancers12051314