The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC)


Abstract
:1. Introduction
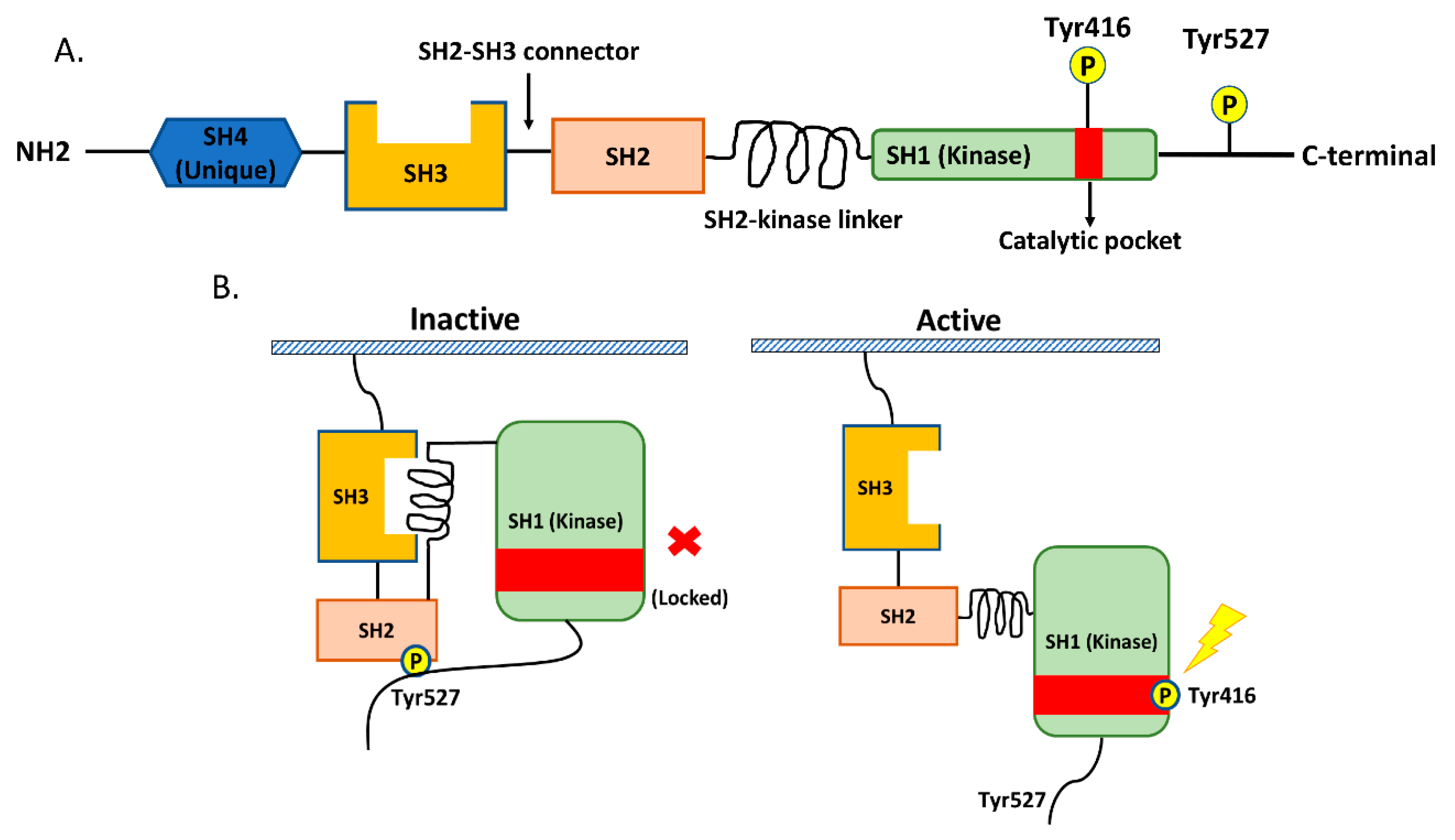
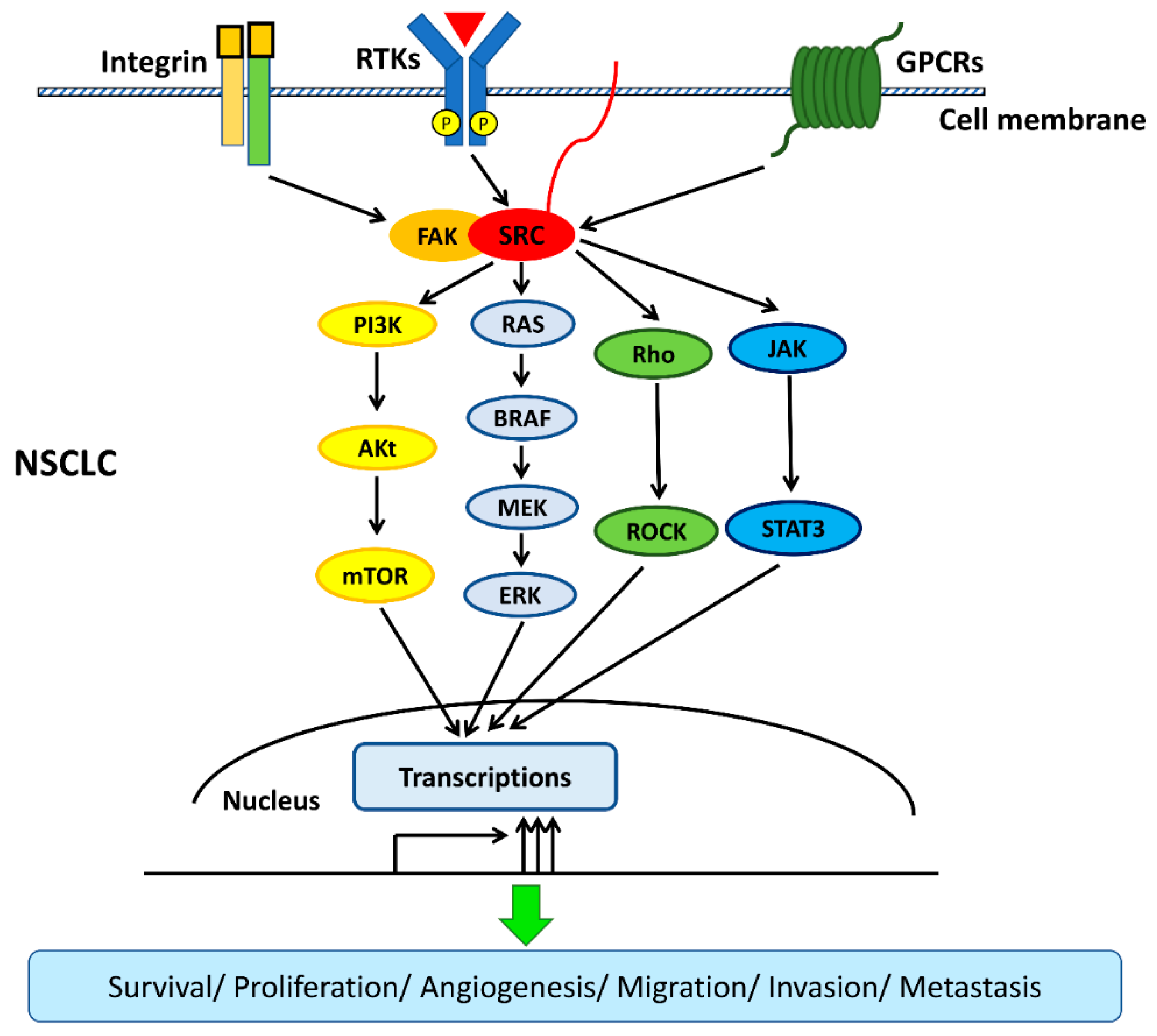
2. Src in NSCLC
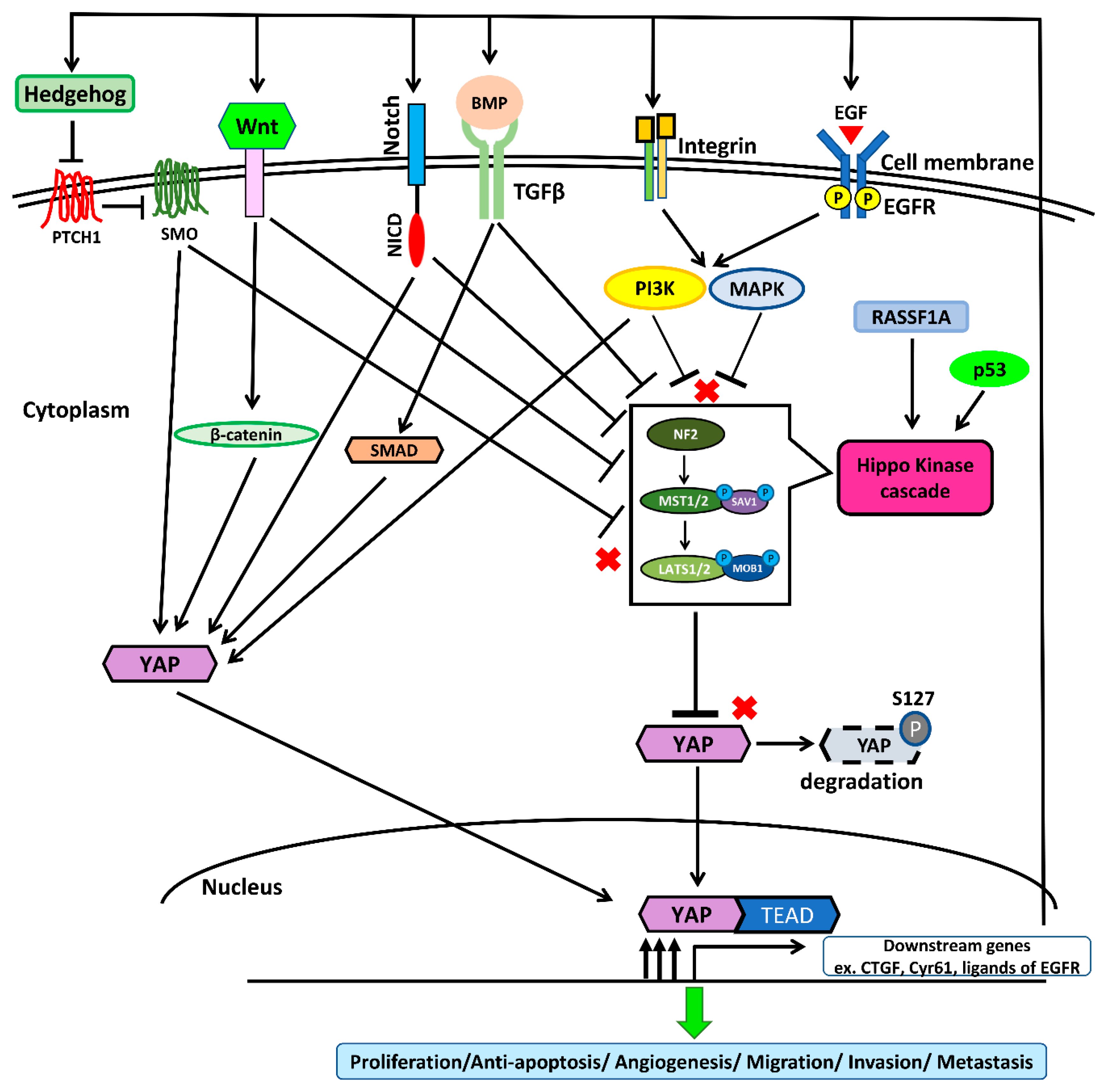
3. Hippo/YAP Singing Pathway in NSCLC
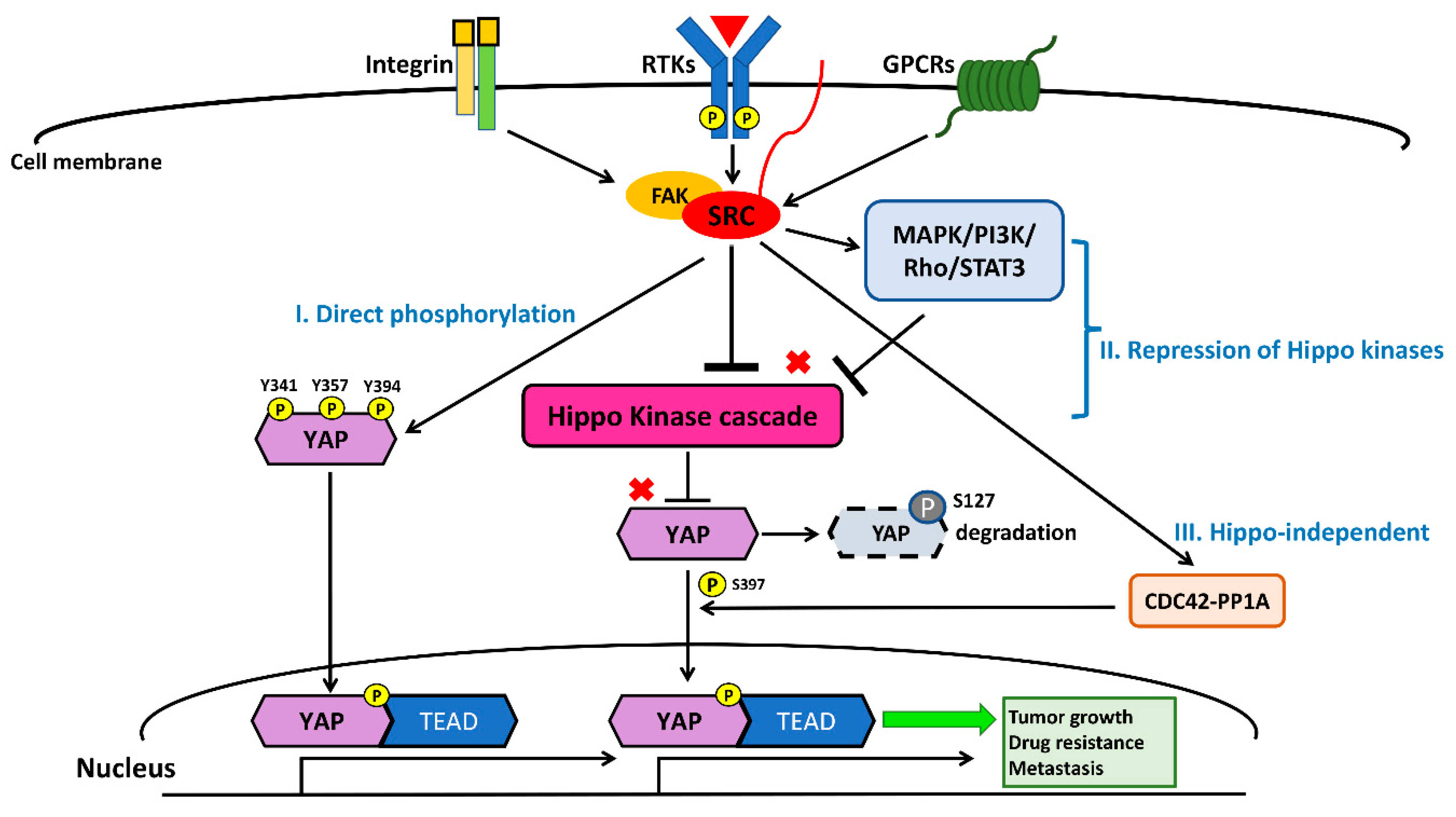
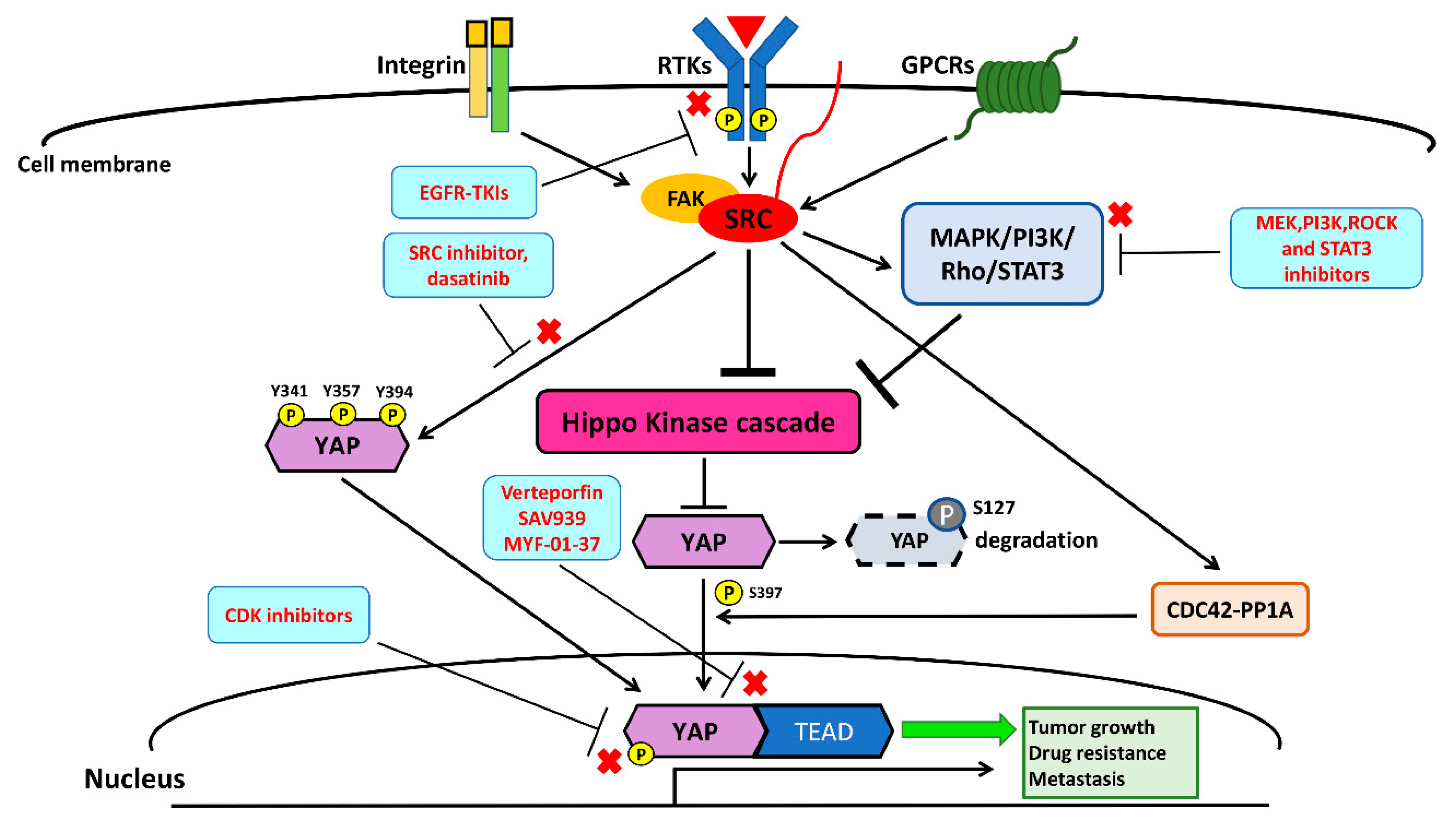
4. Crosstalk between the Src and Hippo/YAP Signaling Pathways
5. Future Perspectives: Potential Therapies Targeting Src–YAP in NSCLC
5.1. Src Inhibitors—Dasatinib, Bosutinib, and Saracatinib
5.2. Inhibition of Src Downstream Intracellular Signaling Pathways
5.3. Disruption of the YAP–TEAD Complex
5.4. Cyclin-Dependent Kinase 1,7,9
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McIntyre, A.; Ganti, A.K. Lung cancer-A global perspective. J. Surg. Oncol. 2017, 115, 550–554. [Google Scholar] [CrossRef]
- Cao, M.; Chen, W. Epidemiology of lung cancer in China. Thorac. Cancer 2019, 10, 3–7. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Bulotta, A.; Ducceschi, M.; Viganò, M.G.; Brioschi, E.; Corti, F.; Gianni, L.; Gregorc, V. Historical Evolution of Second-Line Therapy in Non-small-cell Lung Cancer. Front Med. 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saber, A.; van der Wekken, A.; Hiltermann, T.J.; Kok, K.; Van den Berg, A.; Groen, H. Genomic aberrations guiding treatment of non-small-cell lung cancer patients. Cancer Treat. Commun. 2015, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Ogunleyea, F.; Blankenshipa, L.; Millisorb, V.; Anderson, J.; Jaiyesimi, I. Programmed cell death-1/Programmed cell death ligand-1(PD-1/PD-L1) inhibitors, heralding a new era of immunotherapy in the management of advanced Non-small-cell Lung Cancer (NSCLC). Cancer Treat. Res. Commun. 2017, 12, 6–13. [Google Scholar] [CrossRef]
- Singal, G.; Miller, P.G.; Agarwala, V.; Li, G.; Kaushik, G.; Backenroth, D.; Gossai, A.; Frampton, G.M.; Torres, A.Z.; Lehnert, E.M.; et al. Association of Patient Characteristics and Tumor Genomics With Clinical Outcomes Among Patients With Non-Small Cell Lung Cancer Using a Clinicogenomic Database. JAMA 2019, 321, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small-cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Hsu, P.C.; Yang, C.T.; Jablons, D.M.; You, L. The Role of Yes-Associated Protein (YAP) in Regulating Programmed Death-Ligand 1 (PD-L1) in Thoracic Cancer. Biomedicines 2018, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, F.; Rocco, D.; Vitiello, F.; De Palma, R.; Guerra, G.; De Luca, A.; Navani, N.; Bianco, A. Immune Checkpoint Blockade for Advanced NSCLC: A New Landscape for Elderly Patients. Int. J. Mol. Sci. 2019, 20, 2258. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.C.; Jablons, D.M.; Yang, C.T.; You, L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-small-cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, N.H.; Schneider, B.J.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D.; et al. Therapy for Stage IV Non-Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. J. Clin. Oncol. 2020, 38, 1608–1632. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Liu, W.; Kovacevic, Z.; Peng, Z.; Jin, R.; Wang, P.; Yue, F.; Zheng, M.; Huang, M.L.; Jansson, P.J.; Richardson, V.; et al. The molecular effect of metastasis suppressors on Src signaling and tumorigenesis: New therapeutic targets. Oncotarget 2015, 6, 35522–35541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Teng, L. YAP/TAZ for cancer therapy: Opportunities and challenges (review). Int. J. Oncol. 2015, 46, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Janse van Rensburg, H.J.; Yang, X. The roles of the Hippo pathway in cancer metastasis. Cell Signal. 2016, 28, 1761–1772. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [Green Version]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Okutani, D.; Lodyga, M.; Han, B.; Liu, M. Src Protein Tyrosine Kinase Family and Acute Inflammatory Responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L129–L141. [Google Scholar] [CrossRef] [PubMed]
- Li, L.F.; Kao, K.C.; Liu, Y.Y.; Lin, C.W.; Chen, N.H.; Lee, C.S.; Wang, C.W.; Yang, C.T. Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the Src pathway. J. Cell Mol. Med. 2017, 21, 2937–2949. [Google Scholar] [CrossRef]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: Possible combinations in solid tumors. Clin. Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Kuo, F.C.; Wang, C.L.; Kuo, C.H.; Wang, S.S.; Chen, C.Y.; Huang, Y.B.; Cheng, K.H.; Yokoyama, K.K.; Chen, C.L.; et al. Suppression of IL-8-Src signalling axis by 17β-estradiol inhibits human mesenchymal stem cells-mediated gastric cancer invasion. J. Cell Mol. Med. 2016, 20, 962–972. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Merchant, N.B. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front Biosci. 2012, 17, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Gargalionis, A.N.; Karamouzis, M.V.; Papavassiliou, A.G. The molecular rationale of Src inhibition in colorectal carcinomas. Int. J. Cancer 2014, 134, 2019–2029. [Google Scholar] [CrossRef]
- He, P.; Wu, W.; Wang, H.; Liao, K.; Zhang, W.; Xiong, G.; Wu, F.; Meng, G.; Yang, K. Co-expression of Rho guanine nucleotide exchange factor 5 and Src associates with poor prognosis of patients with resected non-small-cell lung cancer. Oncol. Rep. 2013, 30, 2864–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuranami, S.; Yokobori, T.; Mogi, A.; Altan, B.; Yajima, T.; Onozato, R.; Azuma, Y.; Iijima, M.; Kosaka, T.; Kuwano, H. Src kinase-associated phosphoprotein2 expression is associated with poor prognosis in non-small-cell lung cancer. Anticancer Res. 2015, 35, 2411–2415. [Google Scholar] [PubMed]
- Gu, Z.; Fang, X.; Li, C.; Chen, C.; Liang, G.; Zheng, X.; Fan, Q. Increased PTPRA expression leads to poor prognosis through c-Src activation and G1 phase progression in squamous cell lung cancer. Int. J. Oncol. 2017, 51, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liang, Y.; Chan, Q.; Jiang, L.; Dong, J. CX3CL1 promotes lung cancer cell migration and invasion via the Src/focal adhesion kinase signaling pathway. Oncol. Rep. 2019, 41, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Soulières, D.; Moecks, J.; Bara, I.; Mok, T.; Klughammer, B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J. Cell. Mol. Med. 2014, 18, 1519–1539. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small-cell lung cancer: Perplexity and solution (Review). Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Suzawa, K.; Jordan, E.; Zehir, A.; Ni, A.; Kim, R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin. Cancer Res. 2018, 24, 3108–3118. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. 2017, 77, 2990–3000. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Cardona, A.F.; Bracht, J.W.P.; Aldeguer, E.; Drozdowskyj, A.; Fernandez-Bruno, M.; Chaib, I.; Berenguer, J.; Santarpia, M.; Ito, M.; et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): Novel targets in EGFR-mutation positive non-small-cell lung cancer (NSCLC). EBioMedicine 2019, 39, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Fan, P.D.; Narzisi, G.; Jayaprakash, A.D.; Venturini, E.; Robine, N.; Smibert, P.; Germer, S.; Yu, H.A.; Jordan, E.J.; Paik, P.K.; et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc. Natl. Acad. Sci. USA 2018, 115, E6030–E6038. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Xu, L.F.; Zhang, J.; Kong, S.Y.; Wu, M.; Lao, Y.Z.; Zhou, H.; Zhang, L.; Xu, H. SRC and MEK Co-inhibition Synergistically Enhances the Anti-tumor Effect in Both Non-small-cell Lung Cancer (NSCLC) and Erlotinib-Resistant NSCLC. Front Oncol. 2019, 9, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Huang, T.; Lin, C.Y.; Wu, J.; Fan, B.M.; Bian, Z.X. Exploiting cancer’s phenotypic guise against itself: Targeting ectopically expressed peptide G-protein coupled receptors for lung cancer therapy. Oncotarget 2017, 8, 104615–104637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Xu, X.; Wu, M.; Guan, Z.; Su, X.; Chen, S.; Wang, H.; Teng, L. GPRC5A: An Emerging Biomarker in Human Cancer. BioMed Res. Int. 2018, 2018, 1823726. [Google Scholar] [CrossRef] [PubMed]
- Cisowski, J.; O’Callaghan, K.; Kuliopulos, A.; Yang, J.; Nguyen, N.; Deng, Q.; Agarwal, A. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol. 2011, 179, 513–523. [Google Scholar] [CrossRef]
- Covic, L.; Kuliopulos, A. Protease-Activated Receptor 1 as Therapeutic Target in Breast, Lung, and Ovarian Cancer: Pepducin Approach. Int. J. Mol. Sci. 2018, 19, 2237. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Schaeuble, K.; Kindinger, I.; Impellizzieri, D.; Krueger, W.A.; Hauck, C.R.; Boyman, O.; Legler, D.F. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity 2016, 44, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Förster, R.; Worbs, T.; Werth, K. A 4-midable Connection: CCR7 Tetramers Link GPCR to Src Kinase Signaling. Immunity 2016, 44, 9–11. [Google Scholar] [CrossRef]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef]
- Moody, T.W.; Nuche-Berenguer, B.; Moreno, P.; Jensen, R.T. CI-988 Inhibits EGFR Transactivation and Proliferation Caused by Addition of CCK/Gastrin to Lung Cancer Cells. J. Mol. Neurosci. 2015, 56, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.M.; Yang, C.J.; Hsu, Y.L.; Wu, L.Y.; Tsai, Y.C.; Hung, J.Y.; Lien, C.T.; Huang, M.S.; Kuo, P.L. Glabridin inhibits migration, invasion, and angiogenesis of human non-small-cell lung cancer A549 cells by inhibiting the FAK/rho signaling pathway. Integr. Cancer Ther. 2011, 10, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Umelo, I.A.; Wever, O.D.; Kronenberger, P.; Noor, A.; Teugels, E.; Chen, G.; Bracke, M.; Grève, J.D. Combined inhibition of rho-associated protein kinase and EGFR suppresses the invasive phenotype in EGFR-dependent lung cancer cells. Lung Cancer 2015, 90, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodera, K.; Sakurada, A.; Notsuda, H.; Watanabe, T.; Matsuda, Y.; Noda, M.; Endo, C.; Okada, Y. Growth inhibition of KRAS- and EGFR-mutant lung adenocarcinoma by cosuppression of STAT3 and the SRC/ARHGAP35 axis. Oncol. Rep. 2018, 40, 1761–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramburu, A.; Zudaire, I.; Pajares, M.J.; Agorreta, J.; Orta, A.; Lozano, M.D.; Gúrpide, A.; Gómez-Román, J.; Martinez-Climent, J.A.; Jassem, J.; et al. Combined clinical and genomic signatures for the prognosis of early stage non-small-cell lung cancer based on gene copy number alterations. BMC Genom. 2015, 16, 752. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bértolo, C.; Sainz, C.; Lavín, A.; Remírez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer. 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Chen, L.; Feng, P.; Peng, A.; Qiu, X.; Zhu, X.; He, S.; Zhou, D. cAMP response element-binding protein and Yes-associated protein form a feedback loop that promotes neurite outgrowth. J. Cell. Mol. Med. 2018, 22, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Huo, X.; Zhang, Q.; Liu, A.M.; Tang, C.; Gong, Y.; Bian, J.; Luk, J.M.; Xu, Z.; Chen, J. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol. Rep. 2013, 29, 840–846. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, B.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Liu, S.; Zhang, W.Q.; Yang, Y.L.; Hang, P.; Wang, H.; Cheng, L.; Hsu, P.C.; Wang, Y.C.; Xu, Z.; et al. YAP1 regulates ABCG2 and cancer cell side population in human lung cancer cells. Oncotarget 2017, 8, 4096–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, S.; Chen, J.; Gan, C.; Chen, D.; Li, Y.; Wen, J.; Kremerskothen, J.; Chen, S.; Zhang, J.; et al. WWC2 is an independent prognostic factor and prevents invasion via Hippo signalling in hepatocellular carcinoma. J. Cell. Mol. Med. 2017, 21, 3718–3729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Q.; Dai, Y.Y.; Hsu, P.C.; Wang, H.; Cheng, L.; Yang, Y.L.; Wang, Y.C.; Xu, Z.; Liu, S.; Chan, G.; et al. Targeting YAP in malignant pleural mesothelioma. J. Cell. Mol. Med. 2017, 21, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zeng, M.; Zhu, H.; Chen, X.; Weng, Z.; Li, S. Emerging role of Hippo signalling pathway in bladder cancer. J. Cell. Mol. Med. 2018, 22, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.Y.; Luo, Q.Q.; Xu, Y.H.; Tang, N.W.; Niu, X.M.; Li, Z.M.; Shen, S.P.; Lu, S.; Chen, Z.W. 17-AAG suppresses growth and invasion of lung adenocarcinoma cells via regulation of the LATS1/YAP pathway. J. Cell. Mol. Med. 2015, 19, 651–663. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of Yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Xie, L.X.; Zhang, X.Y.; Hu, P.; Long, M.F.; Xiong, F.; Huang, J.; Ye, X.Q. Role of YAP in lung cancer resistance to cisplatin. Oncol. Lett. 2018, 16, 3949–3954. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Wargo, J.A.; Bivona, T.G. YAP in MAPK pathway targeted therapy resistance. Cell Cycle 2015, 14, 1765–1766. [Google Scholar] [CrossRef]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small-cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, M.; Kleinberg, L.; Halvorsen, A.R.; Helland, A.; Brustugun, O.T. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 2017, 8, 497–504. [Google Scholar] [PubMed] [Green Version]
- Jin, D.; Wu, Y.; Shao, C.; Gao, Y.; Wang, D.; Guo, J. Norcantharidin reverses cisplatin resistance and inhibits the epithelial mesenchymal transition of human non-small lung cancer cells by regulating the YAP pathway. Oncol. Rep. 2018, 40, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazières, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. RASSF1A Suppresses the Invasion and Metastatic Potential of Human Non-small-cell Lung Cancer Cells by Inhibiting YAP Activation through the GEF-H1/RhoB Pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.C.; Miao, J.; Huang, Z.; Yang, Y.L.; Xu, Z.; You, J.; Dai, Y.; Yeh, C.C.; Chan, G.; Liu, S.; et al. Inhibition of yes-associated protein suppresses brain metastasis of human lung adenocarcinoma in a murine model. J. Cell. Mol. Med. 2018, 22, 3073–3085. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; Tian, B.; Yang, Y.L.; Wang, Y.C.; Liu, S.; Urisman, A.; Yang, C.T.; Xu, Z.; Jablons, D.M.; You, L. Cucurbitacin E inhibits the Yes-associated protein signaling pathway and suppresses brain metastasis of human non-small cell lung cancer in a murine model. Oncol. Rep. 2019, 42, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Taha, Z.; Janse van Rensburg, H.J.; Yang, X. The Hippo Pathway: Immunity and Cancer. Cancers 2018, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.C.; Miao, J.; Wang, Y.C.; Zhang, W.Q.; Yang, Y.L.; Wang, C.W.; Yang, C.T.; Huang, Z.; You, J.; Xu, Z.; et al. Inhibition of yes-associated protein down-regulates PD-L1 (CD274) expression in human malignant pleural mesothelioma. J. Cell. Mol. Med. 2018, 22, 3139–3148. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.T.; Tan, E.H.; Zhai, W.; Hillmer, A.M.; Tam, W.L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, H.; Chung, J.Y.; Ylaya, K.; Conway, C.; Takikita, M.; Fukuoka, J.; Doki, Y.; Hanaoka, J.; Hewitt, S.M. Profiling of phospho-AKT, phospho-mTOR, phospho-MAPK and EGFR in non-small-cell lung cancer. J. Histochem. Cytochem. 2014, 62, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhou, H.; Liu, Y.; Huang, J.; Liu, W.; Zhang, Q.; Tang, Q.; Sheng, F.; Li, G.; Zhang, R. ROCK1 promotes migration and invasion of non-small-cell lung cancer cells through the PTEN/PI3K/FAK pathway. Int. J. Oncol. 2019, 55, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Liu, B.; Xiong, H.; Wu, F.; Hu, C.; Liu, P. Trans-3,5,4′-trimethoxystilbene reduced gefitinib resistance in NSCLCs via suppressing MAPK/Akt/Bcl-2 pathway by upregulation of miR-345 and miR-498. J. Cell Mol. Med. 2019, 23, 2431–2441. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Gu, C.; Jeong, K.J.; Zhang, D.; Guo, W.; Lu, Y.; Ju, Z.; Panupinthu, N.; Yang, J.Y.; Gagea, M.M.; et al. YAP/TAZ-Mediated Upregulation of GAB2 Leads to Increased Sensitivity to Growth Factor-Induced Activation of the PI3K Pathway. Cancer Res. 2017, 77, 1637–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Kang, H.N.; Pyo, K.H.; Shin, E.J.; et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK-rearranged lung cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef]
- Chaib, I.; Karachaliou, N.; Pilotto, S.; Codony Servat, J.; Cai, X.; Li, X.; Drozdowskyj, A.; Servat, C.C.; Yang, J.; Hu, C.; et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) Pathway in EGFR-Mutant NSCLC. J. Natl. Cancer Inst. 2017, 109, djx014. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104.e12–122.e12. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, H.; Zhang, T.; Jiang, L.; Li, X.; Liu, S.; Zheng, C.; Yan, G.; Chen, W.; Ning, Y.; et al. miR-25 Promotes Cell Proliferation, Migration, and Invasion of Non-Small-Cell Lung Cancer by Targeting the LATS2/YAP Signaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 9719723. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.M.; Wu, X.; Xian, X.; Wang, L.Y.; Zhu, L.Y.; Sun, H.Y.; Yang, L.; Liu, W.X. Calcitonin gene-related peptide inhibits angiotensin II-induced NADPH oxidase-dependent ROS via the Src/STAT3 signalling pathway. J. Cell Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.Y.; Tang, S.J.; Wu, Y.C.; Yang, K.C.; Huang, H.J.; Sun, G.H.; Sun, K.H. Platinum-based combination chemotherapy triggers cancer cell death through induction of BNIP3 and ROS, but not autophagy. J. Cell Mol. Med. 2020, 24, 1993–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, W.; Pan, Y.; Gao, Y.; Deng, L.; Li, F.; Li, F.; Ma, X.; Hou, S.; Xu, J.; et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63-GPX2 Axis and ROS Accumulation. Cancer Res. 2017, 77, 5769–5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, D.H.; Luu, T.T.; Kim, D.; An, Y.J.; Park, S.; Park, H.J.; Lee, S.K. BMP4 Upregulation Is Associated with Acquired Drug Resistance and Fatty Acid Metabolism in EGFR-Mutant Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 12, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, M.; Zhang, W.; Li, Y.; Zhu, J.; Zhang, X.; Zhao, L.; Zhu, S.; Chen, B. Downregulation of BarH-like homeobox 2 promotes cell proliferation, migration and aerobic glycolysis through Wnt/β-catenin signaling, and predicts a poor prognosis in non-small cell lung carcinoma. Thorac. Cancer 2018, 9, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, Y.; Mao, J.H.; Hsieh, D.; Kim, I.J.; Hu, L.M.; Xu, Z.; Long, H.; Jablons, D.M.; You, L. Inhibition of CK2α down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS ONE 2012, 7, e38996. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2α down-regulates Notch1 signalling in lung cancer cells. Version 2. J. Cell Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Li, Z.H.; Lei, L.; Liu, C.C.; Wang, Z.; Fei, L.R.; Yang, M.Q.; Huang, W.J.; Xu, H.T. FAM83A Promotes Lung Cancer Progression by Regulating the Wnt and Hippo Signaling Pathways and Indicates Poor Prognosis. Front Oncol. 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isago, H.; Mitani, A.; Mikami, Y.; Horie, M.; Urushiyama, H.; Hamamoto, R.; Terasaki, Y.; Nagase, T. Epithelial Expression of YAP and TAZ Is Sequentially Required in Lung Development. Am. J. Respir. Cell Mol. Biol. 2020, 62, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A.; et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife 2018, 7, e31037. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Avantaggiati, M.L.; Nemazanyy, I.; Di Poto, C.; Yang, Y.; Pende, M.; Gibney, G.T.; Ressom, H.W.; Field, J.; Atkins, M.B.; et al. YAP/TAZ Inhibition Induces Metabolic and Signaling Rewiring Resulting in Targetable Vulnerabilities in NF2-Deficient Tumor Cells. Dev. Cell 2019, 49, 425.e9–443.e9. [Google Scholar] [CrossRef]
- Felley-Bosco, E.; Stahel, R. Hippo/YAP pathway for targeted therapy. Transl. Lung Cancer Res. 2014, 3, 75–83. [Google Scholar]
- Malik, S.A.; Khan, M.S.; Dar, M.; Hussain, M.U.; Shah, M.A.; Shafi, S.M.; Mudassar, S. Molecular Alterations and Expression Dynamics of LATS1 and LATS2 Genes in Non-Small-Cell Lung Carcinoma. Pathol. Oncol. Res. 2018, 24, 207–214. [Google Scholar] [CrossRef]
- Raj, N.; Bam, R. Reciprocal Crosstalk Between YAP1/Hippo Pathway and the p53 Family Proteins: Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.; Wang, D.Z.; Pu, W.T. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Fu, J.; Zhou, M.; Xiao, L.; Feng, X.; Chen, H.; Huang, W. Activated Hippo/Yes-Associated Protein Pathway Promotes Cell Proliferation and Anti-apoptosis in Endometrial Stromal Cells of Endometriosis. J. Clin. Endocrinol. Metab. 2016, 101, 1552–1561. [Google Scholar] [CrossRef]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.P.; Rundsten, C.F.; et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35.e7–49.e7. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ege, N.; Dowbaj, A.M.; Jiang, M.; Howell, M.; Hooper, S.; Foster, C.; Jenkins, R.P.; Sahai, E. Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018, 6, 692–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Jung, S.Y.; Song, K.H.; Park, J.I.; Ahn, J.; Kim, E.H.; Park, J.K.; Hwang, S.G.; Woo, H.J.; Song, J.Y. A new FGFR inhibitor disrupts the TGF-β1-induced fibrotic process. J. Cell Mol. Med. 2020, 24, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Tang, J.; Overstreet, J.M.; Anorga, S.; Lian, F.; Arnouk, A.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 2019, 33, 9797–9810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Choi, H.I.; Park, J.S.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. FASEB J. 2019, 33, 11109–11122. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/bies.201900162 (accessed on 26 May 2020).
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [Green Version]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.H.; Arron, S.T.; Vasioukhin, V. αE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Smoot, R.L.; Werneburg, N.W.; Sugihara, T.; Hernandez, M.C.; Yang, L.; Mehner, C.; Graham, R.P.; Bronk, S.F.; Truty, M.J.; Gores, G.J. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J. Cell Biochem. 2018, 119, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Y.; Ji, X.; Cao, X.; Dai, X.; Xu, L.; Zhao, H.; Guo, X.; Yan, H.; Zhang, H.; Zhu, C.; et al. Src Inhibits the Hippo Tumor Suppressor Pathway Through Tyrosine Phosphorylation of Lats1. Cancer Res. 2017, 77, 4868–4880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, T.; Charindra, D.; Shrestha, M.; Umehara, H.; Ogawa, I.; Miyauchi, M.; Takata, T. Tissue inhibitor of metalloproteinase-1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene 2018, 37, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Moroishi, T.; de Jong, P.R.; Krawczyk, M.; Grebbin, B.M.; Luo, H.; Xu, R.H.; Golob-Schwarzl, N.; Schweiger, C.; Wang, K.; et al. YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Imada, S.; Murata, Y.; Kotani, T.; Hatano, M.; Sun, C.; Konno, T.; Park, J.H.; Kitamura, Y.; Saito, Y.; Ohdan, H.; et al. Role of Src Family Kinases in Regulation of Intestinal Epithelial Homeostasis. Mol. Cell Biol. 2016, 36, 2811–2823. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt Phosphorylates the Yes-Associated Protein, YAP, to Induce Interaction with 14-3-3 and Attenuation of p73-Mediated Apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Hu, J.K.; Du, W.; Shelton, S.J.; Oldham, M.C.; DiPersio, C.M.; Klein, O.D. An FAK-YAP-mTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell 2017, 21, 91.e6–106.e6. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.S.; Yap, S.H.; Phuah, N.H.; In, L.L.; Hasima, N. MicroRNAs associated with tumour migration, invasion and angiogenic properties in A549 and SK-Lu1 human lung adenocarcinoma cells. Lung Cancer 2014, 83, 154–162. [Google Scholar] [CrossRef]
- Rao, G.; Kim, I.K.; Conforti, F.; Liu, J.; Zhang, Y.W.; Giaccone, G. Dasatinib sensitises KRAS-mutant cancer cells to mitogen-activated protein kinase kinase inhibitor via inhibition of TAZ activity. Eur. J. Cancer 2018, 99, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.; Ditzel, H.J. The potential of Src inhibitors. Aging 2015, 7, 734–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, R.; Hsyu, P.H. Clinical Pharmacokinetics and Pharmacodynamics of Bosutinib. Clin Pharmacokinet. 2016, 55, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Brinda, B.; Khan, I.; Parkin, B.; Konig, H. The rocky road to personalized medicine in acute myeloid leukaemia. J. Cell. Mol. Med. 2018, 22, 1411–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, X.; Tang, B.; Liu, H.; Zhang, M.; Wang, Y.; Ping, F.; Ding, J.; Shen, A.; Geng, M. A tightly controlled Src-YAP signaling axis determines therapeutic response to dasatinib in renal cell carcinoma. Theranostics 2018, 8, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Gold, K.A.; Lee, J.J.; Harun, N.; Tang, X.; Price, J.; Kawedia, J.D.; Tran, H.T.; Erasmus, J.J.; Blumenschein, G.R.; William, W.N.; et al. A phase I/II study combining erlotinib and dasatinib for non-small-cell lung cancer. Oncologist 2014, 19, 1040–1041. [Google Scholar] [CrossRef] [Green Version]
- Laurie, S.A.; Goss, G.D.; Shepherd, F.A.; Reaume, M.N.; Nicholas, G.; Philip, L.; Wang, L.; Schwock, J.; Hirsh, V.; Oza, A.; et al. A phase II trial of saracatinib, an inhibitor of src kinases, in previously-treated advanced non-small-cell lung cancer: The princess margaret hospital phase II consortium. Clin. Lung Cancer 2014, 15, 52–57. [Google Scholar] [CrossRef]
- Slemmons, K.K.; Yeung, C.; Baumgart, J.T.; Martinez Juarez, J.O.; McCalla, A.; Helman, L.J. Targeting Hippo-dependent and Hippo-independent YAP1 signaling for the treatment of childhood rhabdomyosarcoma. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Strudel, M.; Ascierto, P.A. MEK Inhibitors in the Treatment of Metastatic Melanoma and Solid Tumors. Am. J. Clin. Dermatol. 2017, 18, 745–754. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Baik, C.S.; Myall, N.J.; Wakelee, H.A. Targeting BRAF-Mutant Non-small-cell Lung Cancer: From Molecular Profiling to Rationally Designed Therapy. Oncologist 2017, 22, 786–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinchcombe, T.E.; Johnson, G.L. MEK inhibition in non-small-cell lung cancer. Lung Cancer 2014, 86, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Giaccone, G. MEK inhibitors under development for treatment of non-small-cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Yamamoto, H.; Sakaguchi, M.; Shien, K.; Tomida, S.; Shien, T.; Ikeda, H.; Hatono, M.; Torigoe, H.; Namba, K.; et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small-cell lung cancer. Cancer Sci. 2018, 109, 3183–3196. [Google Scholar] [CrossRef] [PubMed]
- Muranen, T.; Selfors, L.M.; Hwang, J.; Gallegos, L.L.; Coloff, J.L.; Thoreen, C.C.; Kang, S.A.; Sabatini, D.M.; Mills, G.B.; Brugge, J.S. ERK and p38 MAPK Activities Determine Sensitivity to PI3K/mTOR Inhibition via Regulation of MYC and YAP. Cancer Res. 2016, 76, 7168–7180. [Google Scholar] [CrossRef] [Green Version]
- Woodard, G.A.; Yang, Y.L.; You, L.; Jablons, D.M. Drug development against the hippo pathway in mesothelioma. Transl. Lung Cancer Res. 2017, 6, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Lin, F.; Wu, W.; Liu, Y.; Huang, W. Verteporfin inhibits YAP-induced bladder cancer cell growth and invasion via Hippo signaling pathway. Int. J. Med. Sci. 2018, 15, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Chen, Y.; Li, X.; Yang, R.; Zhang, L.; Huangfu, L.; Zheng, N.; Zhao, X.; Lv, L.; Hong, Y.; et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co-factor TEAD. Cell Death Dis. 2018, 9, 464. [Google Scholar] [CrossRef] [Green Version]
- Nowak-Sliwinska, P.; Weiss, A.; van Beijnum, J.R.; Wong, T.J.; Ballini, J.P.; Lovisa, B.; van den Bergh, H.; Griffioen, A.W. Angiostatic kinase inhibitors to sustain photodynamic angio-occlusion. J. Cell Mol. Med. 2012, 16, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, S.P.; Adelman, R.A. Profile of verteporfin and its potential for the treatment of central serous chorioretinopathy. Clin. Ophthalmol. 2013, 7, 1867–1875. [Google Scholar] [PubMed] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, L.; Liu, M.; Chong, R.; Ding, S.J.; Chen, Y.; Dong, J. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013, 73, 6722–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Khanal, P.; Savage, P.; She, Y.M.; Cyr, T.D.; Yang, X. YAP-induced resistance of cancer cells to antitubulin drugs is modulated by a Hippo-independent pathway. Cancer Res. 2014, 74, 4493–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Kyoyama, H.; Liu, L.; Chan, G.; Wang, Y.; Urisman, A.; Yang, Y.L.; Liu, S.; Xu, Z.; Bin, H.; et al. Inhibition of cyclin-dependent kinase 7 down-regulates yes-associated protein expression in mesothelioma cells. J. Cell Mol. Med. 2020, 24, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018, 175, 1244–1258. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.J.; Nemunaitis, J.; Joy, A.A.; Martin, J.C.; Jou, Y.M.; Zhang, D.; Statkevich, P.4.; Yao, S.L.; Zhu, Y.; Zhou, H.; et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer 2014, 83, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Alternation of Src | Primary Mutation of NSCLC | Resistance to Target Therapy (Primary/Secondary) | Targeted Therapy Resistance | Type of Study | Reference |
|---|---|---|---|---|---|
| Overexpression of SHP2 | EGFR mutation L858R/exon 19 deletion/G719X/L861Q | Primary | Erlotinib, Gefitinib, Afatinib | Clinical analysis | [39] |
| YES1 amplification | EGFR mutation L858R/exon 19 deletion | Secondary | Erlotinib, Afatinib | Clinical analysis | [37] |
| YES1 amplification | EGFR mutation Exon 19 deletion/L858R+T790M | Secondary | Osimertinib | Preclinical study | [38] |
| YES1 amplification | EGFR mutation L858R/exon 19 deletion ALK fusion EML4-ALK fusion/HIP1-ALK fusion | Secondary | Erlotinib, Afatinib, Crizotinib, Ceritinib | Clinical analysis | [40] |
| YAP Alternation | Primary NSCLC Mutation | Resistance to Target Therapy (Primary/Secondary) | Targeted Therapy Resistance | Type of Study | Reference |
|---|---|---|---|---|---|
| YAP overexpression | EGFR mutation Exon 19 deletion/L858R+T790M | Primary/secondary | Erlotinib | Preclinical study | [71] |
| YAP overexpression | EGFR mutation L858R/exon 19 deletion/T790M | Primary/secondary | Erlotinib, Gefitinib, Afatinib, Osimertinib | Preclinical study and clinical analysis | [94] |
| YAP overexpression | L858R/exon 19 deletion /G719X/L861Q | Primary | Erlotinib, Gefitinib, Afatinib, Icotinib | Preclinical study and clinical analysis | [97] |
| YAP overexpression | EGFR mutation L858R/exon 19 deletion/T790M | Secondary | Osimertinib, Trametinib | Preclinical study | [98] |
| YAP overexpression | ALK fusion EML4-ALK fusion/ | Secondary | Crizotinib, Ceritinib | Preclinical study | [96] |
| YAP overexpression | BRAF V600E, K-ras | Primary | Vermurafenib, Trametinib | Preclinical study | [89] |
| Target | Drug | Status in NSCLC | Reference |
|---|---|---|---|
| Src | Dasatinib | Phase I/II clinical trial (Completed) NCT00826449 | [148] |
| Bosutinib | Phase I clinical trial (Recruiting) NCT03023319 | [41] | |
| Saracatinib (AZD0530) | Phase II clinical trial (Completed) NCT00638937 | [149] | |
| MAPK | Trametinib | Combined with dabrafenib in BRAF V600E mutation NSCLC (Approved by US FDA) | [152,153] |
| PI3K | Taselisib | Preclinical study | [156] |
| STAT3 | TPCA-1 | Preclinical study | [97] |
| Rho/ROCK | GSK269962A | Preclinical study | [63,158] |
| YAP–TEAD | Verteporfin | Preclinical study | [159,160,161] |
| XAV939 | Preclinical study | [98] | |
| MYF-01-37 | Preclinical study | [98] | |
| CDK1,5,9 | Dinaciclib (MK7965) | Phase II clinical trial (Completed) NCT00732810 | [170,172] |
| CDK7 | THZ1 | Preclinical study | [167,168,169] |
| CDK9 | Seliciclib | Phase II clinical trial (Terminated) NCT00372073 | [170,173] |
| Flavopiridol (alvocidib) | Phase I clinical trial (Terminated) NCT00094978 | [170,171,173] | |
| MC180295 | Preclinical study | [170,171] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.-C.; Yang, C.-T.; Jablons, D.M.; You, L. The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC). Cancers 2020, 12, 1361. https://doi.org/10.3390/cancers12061361
Hsu P-C, Yang C-T, Jablons DM, You L. The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC). Cancers. 2020; 12(6):1361. https://doi.org/10.3390/cancers12061361
Chicago/Turabian StyleHsu, Ping-Chih, Cheng-Ta Yang, David M. Jablons, and Liang You. 2020. "The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC)" Cancers 12, no. 6: 1361. https://doi.org/10.3390/cancers12061361
APA StyleHsu, P. -C., Yang, C. -T., Jablons, D. M., & You, L. (2020). The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC). Cancers, 12(6), 1361. https://doi.org/10.3390/cancers12061361