Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far

 ,
, 
 ,
, 
Abstract
:
1. Introduction
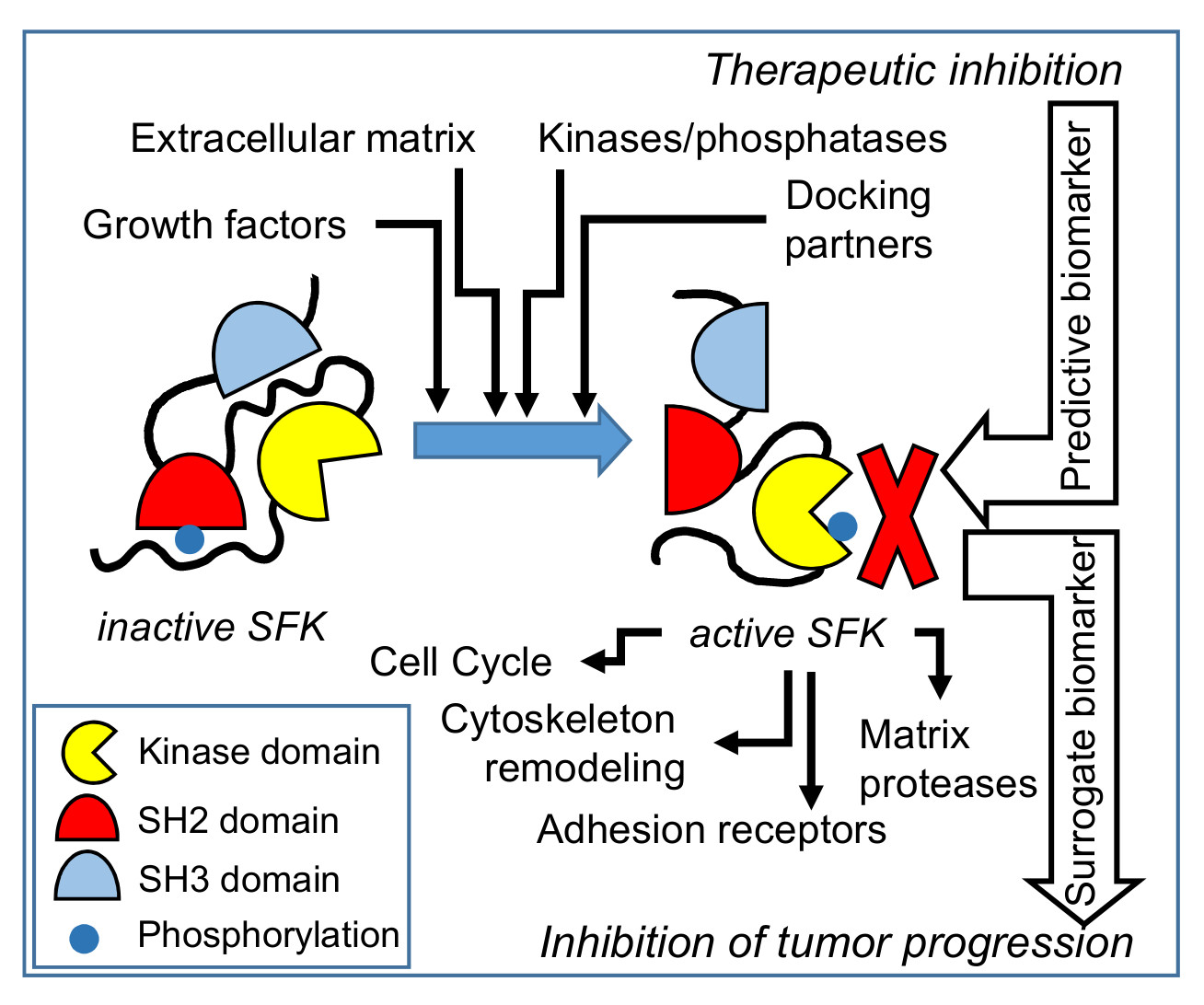
2. SFKs Structure and Regulation
3. SFKs in Cancer Signal Transduction, Migration, and Invasion
4. SFKs and Cancer Progression
4.1. Breast Cancer
4.2. Lung Cancer
4.3. Thyroid Cancer
4.4. Glioblastoma
4.5. Colorectal Cancer
4.6. Pancreatic Cancer
4.7. Prostate Cancer
4.8. Other Cancers
5. Clinical Trials in Advanced Solid Tumors
5.1. Dasatinib
5.2. Saracatinib
5.3. Bosutinib
6. New Src Inhibitors in Clinical Trials
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angelucci, A. Targeting Tyrosine Kinases in Cancer: Lessons for an Effective Targeted Therapy in the Clinic. Cancers 2019, 11, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Rai, K. Personalized Cancer Therapy: YES1 Is the New Kid on the Block. Cancer Res. 2019, 79, 5702–5703. [Google Scholar] [CrossRef] [PubMed]
- Kohmura, N.; Yagi, T.; Tomooka, Y.; Oyanagi, M.; Kominami, R.; Takeda, N.; Chiba, J.; Ikawa, Y.; Aizawa, S. A novel nonreceptor tyrosine kinase, Srm: Cloning and targeted disruption. Mol. Cell. Biol. 1994, 14, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.L.; Vogel, H.; Soriano, P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994, 8, 1999–2007. [Google Scholar] [CrossRef] [Green Version]
- Yim, E.K.; Peng, G.; Dai, H.; Hu, R.; Li, K.; Lu, Y.; Mills, G.B.; Meric-Bernstam, F.; Hennessy, B.T.; Craven, R.J.; et al. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 2009, 15, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Hung, W.S.; Chan, H.H.; Tsai, S.J.; Sun, H.S. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics 2013, 29, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Je, D.W.; Ji, Y.G.; Cho, Y.; Lee, D.H. The inhibition of SRC family kinase suppresses pancreatic cancer cell proliferation, migration, and invasion. Pancreas 2014, 43, 768–776. [Google Scholar] [CrossRef]
- Sudol, M.; Greulich, H.; Newman, L.; Sarkar, A.; Sukegawa, J.; Yamamoto, T. A novel Yes-related kinase, Yrk, is expressed at elevated levels in neural and hematopoietic tissues. Oncogene 1993, 8, 823–831. [Google Scholar]
- Martins-Green, M.; Bixby, J.L.; Yamamoto, T.; Graf, T.; Sudol, M. Tissue specific expression of Yrk kinase: Implications for differentiation and inflammation. Int. J. Biochem. Cell B 2000, 32, 351–364. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.S.; Cartagena-Rivera, A.X.; Chadwick, R.S. c-Src activity is differentially required by cancer cell motility modes. Oncogene 2018, 37, 2104–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Li, X.; Song, N.; Li, A.; Hou, K.; Qu, X.; Che, X.; Liu, Y. Src promotes EGF-induced epithelial-to-mesenchymal transition and migration in gastric cancer cells by upregulating ZEB1 and ZEB2 through AKT. Cell Biol. Int. 2018, 42, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Airik, M.; Krook, M.A.; Pedersen, E.A.; Lawlor, E.R. Micro-Environmental Stress Induces Src-Dependent Activation of Invadopodia and Cell Migration in Ewing Sarcoma. Neoplasia 2016, 18, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngan, E.; Stoletov, K.; Smith, H.W.; Common, J.; Muller, W.J.; Lewis, J.D.; Siegel, P.M. LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun. 2017, 8, 15059. [Google Scholar] [CrossRef]
- Calgani, A.; Vignaroli, G.; Zamperini, C.; Coniglio, F.; Festuccia, C.; Di Cesare, E.; Gravina, G.L.; Mattei, C.; Vitale, F.; Schenone, S.; et al. Suppression of SRC Signaling Is Effective in Reducing Synergy between Glioblastoma and Stromal Cells. Mol. Cancer Ther. 2016, 15, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Delle Monache, S.; Sanita, P.; Calgani, A.; Schenone, S.; Botta, L.; Angelucci, A. Src inhibition potentiates antitumoral effect of paclitaxel by blocking tumor-induced angiogenesis. Exp. Cell Res. 2014, 328, 20–31. [Google Scholar] [CrossRef]
- Liu, S.T.; Pham, H.; Pandol, S.J.; Ptasznik, A. Src as the link between inflammation and cancer. Front. Physiol. 2013, 4, 416. [Google Scholar] [CrossRef] [Green Version]
- Destaing, O.; Sanjay, A.; Itzstein, C.; Horne, W.C.; Toomre, D.; De Camilli, P.; Baron, R. The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol. Biol. Cell 2008, 19, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myoui, A.; Nishimura, R.; Williams, P.J.; Hiraga, T.; Tamura, D.; Michigami, T.; Mundy, G.R.; Yoneda, T. C-SRC tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003, 63, 5028–5033. [Google Scholar] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, K.B.; Bibbins, K.B.; Swedlow, J.R.; Arnaud, M.; Morgan, D.O.; Varmus, H.E. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 1994, 13, 4745–4756. [Google Scholar] [CrossRef]
- Wu, J.C.; Chen, Y.C.; Kuo, C.T.; Wenshin Yu, H.; Chen, Y.Q.; Chiou, A.; Kuo, J.C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5, 18476. [Google Scholar] [CrossRef] [Green Version]
- Paladino, D.; Yue, P.; Furuya, H.; Acoba, J.; Rosser, C.J.; Turkson, J. A novel nuclear Src and p300 signaling axis controls migratory and invasive behavior in pancreatic cancer. Oncotarget 2016, 7, 7253–7267. [Google Scholar] [CrossRef] [Green Version]
- Urciuoli, E.; Coletta, I.; Rizzuto, E.; De Vito, R.; Petrini, S.; D’Oria, V.; Pezzullo, M.; Milano, G.M.; Cozza, R.; Locatelli, F.; et al. Src nuclear localization and its prognostic relevance in human osteosarcoma. J. Cell Physiol. 2018, 233, 1658–1670. [Google Scholar] [CrossRef]
- Amata, I.; Maffei, M.; Pons, M. Phosphorylation of unique domains of Src family kinases. Front. Genet. 2014, 5, 181. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Chai, C.Y.; Yeh, K.T.; Wang, S.N.; Tsai, C.J.; Yeh, Y.T.; Yang, S.F. Crosstalk between activated and inactivated c-Src in hepatocellular carcinoma. Dis. Markers 2011, 30, 325–333. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [Green Version]
- Mustelin, T.; Tasken, K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 2003, 371, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Y.P.; Ia, K.K.; Mulhern, T.D.; Cheng, H.C. Endogenous and synthetic inhibitors of the Src-family protein tyrosine kinases. Biochim. Biophys. Acta 2005, 1754, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Minowa, T.; Hanagata, N.; Kataoka-Hamai, C.; Kaizuka, Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J. Biol. Chem. 2014, 289, 28514–28525. [Google Scholar] [CrossRef] [Green Version]
- Chong, Y.P.; Mulhern, T.D.; Cheng, H.C. C-terminal Src kinase (CSK) and CSK-homologous kinase (CHK)--endogenous negative regulators of Src-family protein kinases. Growth Factors 2005, 23, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, T.E.; Shalloway, D. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell 1987, 49, 65–73. [Google Scholar] [CrossRef]
- Abram, C.L.; Courtneidge, S.A. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res. 2000, 254, 1–13. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 2000, 275, 41439–41446. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Esselman, W.J.; Cole, P.A. Substrate conformational restriction and CD45-catalyzed dephosphorylation of tail tyrosine-phosphorylated Src protein. J. Biol. Chem. 2002, 277, 40428–40433. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, H.L.; Shackelford, D.A.; Hurley, T.R.; Johnson, P.; Hyman, R.; Sefton, B.M.; Trowbridge, I.S. Expression of CD45 alters phosphorylation of the lck-encoded tyrosine protein kinase in murine lymphoma T-cell lines. Proc. Natl. Acad. Sci. USA 1989, 86, 8959–8963. [Google Scholar] [CrossRef] [Green Version]
- Somani, A.K.; Bignon, J.S.; Mills, G.B.; Siminovitch, K.A.; Branch, D.R. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 1997, 272, 21113–21119. [Google Scholar] [CrossRef] [Green Version]
- Chiang, G.G.; Sefton, B.M. Specific dephosphorylation of the Lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 2001, 276, 23173–23178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.; Gamble, J.; Tooze, R.; Higgins, D.; Yang, F.T.; O’Brien, P.C.; Coleman, N.; Pingel, S.; Turner, M.; Alexander, D.R. Development of T-leukaemias in CD45 tyrosine phosphatase-deficient mutant lck mice. EMBO J. 2000, 19, 4644–4654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell 2004, 13, 341–355. [Google Scholar] [CrossRef]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorge, J.D.; Jakymiw, A.; Fujita, D.J. Selected glimpses into the activation and function of Src kinase. Oncogene 2000, 19, 5620–5635. [Google Scholar] [CrossRef] [Green Version]
- Gayrard, C.; Bernaudin, C.; Dejardin, T.; Seiler, C.; Borghi, N. Src- and confinement-dependent FAK activation causes E-cadherin relaxation and beta-catenin activity. J. Cell Biol. 2018, 217, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Miller, D.J.; Guibao, C.D.; Donato, D.M.; Hanks, S.K.; Zheng, J.J. Structural and functional insights into the interaction between the Cas family scaffolding protein p130Cas and the focal adhesion-associated protein paxillin. J. Biol. Chem. 2017, 292, 18281–18289. [Google Scholar] [CrossRef] [Green Version]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef]
- Yeo, M.G.; Oh, H.J.; Cho, H.S.; Chun, J.S.; Marcantonio, E.E.; Song, W.K. Phosphorylation of Ser 21 in Fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J. Cell Physiol. 2011, 226, 236–247. [Google Scholar] [CrossRef]
- Chatterji, T.; Varkaris, A.S.; Parikh, N.U.; Song, J.H.; Cheng, C.J.; Schweppe, R.E.; Alexander, S.; Davis, J.W.; Troncoso, P.; Friedl, P.; et al. Yes-mediated phosphorylation of focal adhesion kinase at tyrosine 861 increases metastatic potential of prostate cancer cells. Oncotarget 2015, 6, 10175–10194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Lorenz, M.; Kempiak, S.; Sarmiento, C.; Coniglio, S.; Symons, M.; Segall, J.; Eddy, R.; Miki, H.; Takenawa, T.; et al. Molecular mechanisms of invadopodium formation: The role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 2005, 168, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.; Yamaguchi, H.; Mader, C.C.; Bravo-Cordero, J.J.; Arias, M.; Chen, X.; Desmarais, V.; van Rheenen, J.; Koleske, A.J.; Condeelis, J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 2009, 186, 571–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Liu, J.; Haudenschild, C.C.; Zhan, X. The role of tyrosine phosphorylation of cortactin in the locomotion of endothelial cells. J. Biol. Chem. 1998, 273, 25770–25776. [Google Scholar] [CrossRef] [Green Version]
- Tehrani, S.; Tomasevic, N.; Weed, S.; Sakowicz, R.; Cooper, J.A. Src phosphorylation of cortactin enhances actin assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 11933–11938. [Google Scholar] [CrossRef] [Green Version]
- Desmarais, V.; Yamaguchi, H.; Oser, M.; Soon, L.; Mouneimne, G.; Sarmiento, C.; Eddy, R.; Condeelis, J. N-WASP and cortactin are involved in invadopodium-dependent chemotaxis to EGF in breast tumor cells. Cell Motil. Cytoskelet. 2009, 66, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Boateng, L.R.; Cortesio, C.L.; Huttenlocher, A. Src-mediated phosphorylation of mammalian Abp1 (DBNL) regulates podosome rosette formation in transformed fibroblasts. J. Cell Sci. 2012, 125, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Artym, V.V.; Zhang, Y.; Seillier-Moiseiwitsch, F.; Yamada, K.M.; Mueller, S.C. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: Defining the stages of invadopodia formation and function. Cancer Res. 2006, 66, 3034–3043. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Feng, R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Merchant, N.B. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front. Biosci. (Landmark Ed) 2012, 17, 2059–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, D.K.; Lee, A.; Lansing, T.J.; Crosby, R.M.; Jung, K.D.; Willard, D.; Luther, M.; Rodriguez, M.; Berman, J.; Gilmer, T.M. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc. Natl. Acad. Sci. USA 1994, 91, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, S.M.; Seo, J.; Hwang, J.T.; Sung, G.J.; Song, J.H.; Jeong, J.H.; Lee, S.H.; Yoon, H.G.; Choi, H.K.; Choi, K.C. EGFR-c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells. Cells 2019, 8, 930. [Google Scholar] [CrossRef] [Green Version]
- Lou, L.; Yu, Z.; Wang, Y.; Wang, S.; Zhao, Y. c-Src inhibitor selectively inhibits triple-negative breast cancer overexpressed Vimentin in vitro and in vivo. Cancer Sci. 2018, 109, 1648–1659. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Nobre, A.R.; Mendes, N.; Almeida, J.; Vieira, A.F.; Sousa, B.; Carvalho, F.A.; Monteiro, J.; Polonia, A.; Fonseca, M.; et al. SRC inhibition prevents P-cadherin mediated signaling and function in basal-like breast cancer cells. Cell Commun. Signal. 2018, 16, 75. [Google Scholar] [CrossRef] [Green Version]
- Olea-Flores, M.; Zuniga-Eulogio, M.; Tacuba-Saavedra, A.; Bueno-Salgado, M.; Sanchez-Carvajal, A.; Vargas-Santiago, Y.; Mendoza-Catalan, M.A.; Perez Salazar, E.; Garcia-Hernandez, A.; Padilla-Benavides, T.; et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells 2019, 8, 1133. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.G.; Yu, Y.; Hou, L.K.; Wang, X.; Zhang, B.; Cao, X.C. FYN promotes breast cancer progression through epithelial-mesenchymal transition. Oncol. Rep. 2016, 36, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, G.; Rangaswami, H.; Jain, S.; Kundu, G.C. Hypoxia regulates cross-talk between Syk and Lck leading to breast cancer progression and angiogenesis. J. Biol. Chem. 2006, 281, 11322–11331. [Google Scholar] [CrossRef] [Green Version]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Cooke, J.; Mirza, H.; Aurrekoetxea-Rodriguez, I.; Vivanco, M.D.M.; Buckley, N.E.; Grigoriadis, A.; Smalley, M.J. Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. Cell Rep. 2018, 25, 3674–3692.e3610. [Google Scholar] [CrossRef] [Green Version]
- Ogunbolude, Y.; Dai, C.; Bagu, E.T.; Goel, R.K.; Miah, S.; MacAusland-Berg, J.; Ng, C.Y.; Chibbar, R.; Napper, S.; Raptis, L.; et al. FRK inhibits breast cancer cell migration and invasion by suppressing epithelial-mesenchymal transition. Oncotarget 2017, 8, 113034–113065. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Annis, M.G.; Hsu, B.E.; Tam, C.E.; Savage, P.; Park, M.; Siegel, P.M. Lyn modulates Claudin-2 expression and is a therapeutic target for breast cancer liver metastasis. Oncotarget 2015, 6, 9476–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.H.; Lin, S.Y.; Wu, Y.S.; Chen, H.W.; Chen, J.J.W. AC-93253 iodide, a novel Src inhibitor, suppresses NSCLC progression by modulating multiple Src-related signaling pathways. J. Hematol. Oncol. 2017, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Nicholes, K.; Bustos, D.; Lin, E.; Song, Q.; Stephan, J.P.; Kirkpatrick, D.S.; Settleman, J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014, 5, 7328–7341. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Peng, W.H.; Wu, C.H.; Chang, Y.M.; Lin, Y.L.; Chang, G.D.; Wu, H.C.; Chen, G.C. PTPN3 suppresses lung cancer cell invasiveness by counteracting Src-mediated DAAM1 activation and actin polymerization. Oncogene 2019, 38, 7002–7016. [Google Scholar] [CrossRef]
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bertolo, C.; Sainz, C.; Lavin, A.; Remirez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899. [Google Scholar] [CrossRef]
- Li, H.Y.; Carr, L.L. Predictive Biomarkers of Response to Src Inhibitors in Lung Cancer. Getting to YES1. Am. J. Respir. Crit. Care Med. 2019, 200, 802–804. [Google Scholar] [CrossRef]
- Beadnell, T.C.; Nassar, K.W.; Rose, M.M.; Clark, E.G.; Danysh, B.P.; Hofmann, M.C.; Pozdeyev, N.; Schweppe, R.E. Src-mediated regulation of the PI3K pathway in advanced papillary and anaplastic thyroid cancer. Oncogenesis 2018, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin. Cancer Res. 2012, 18, 3580–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Stewart, J., Jr.; Olman, M.A.; Klobe, M.R.; Gladson, C.L. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 2003, 278, 39882–39891. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhang, W.; Yang, X.; Wheeler, C.G.; Langford, C.P.; Wu, L.; Filippova, N.; Friedman, G.K.; Ding, Q.; Fathallah-Shaykh, H.M.; et al. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 2014, 45, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef] [Green Version]
- Korinek, V.; Barker, N.; Willert, K.; Molenaar, M.; Roose, J.; Wagenaar, G.; Markman, M.; Lamers, W.; Destree, O.; Clevers, H. Two members of the Tcf family implicated in Wnt/beta-catenin signaling during embryogenesis in the mouse. Mol. Cell. Biol. 1998, 18, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Wang, H.; Zhang, Z.; Zhang, M.; Qu, Y.; Zhao, Z.; Fan, X.; Zhan, Q.; Song, Y.; Yu, C. Downregulation of beta-arrestin 1 suppresses glioblastoma cell malignant progression via inhibition of Src signaling. Exp. Cell Res. 2017, 357, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Vitale, S.; Avizienyte, E.; Brunton, V.G.; Frame, M.C. Focal adhesion kinase is not required for Src-induced formation of invadopodia in KM12C colon cancer cells and can interfere with their assembly. Eur. J. Cell Biol. 2008, 87, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Taulet, N.; DerMardirossian, C.; Bokoch, G.M. c-Src-mediated phosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)-dependent formation of functional invadopodia in human colon cancer cells. Mol. Biol. Cell 2010, 21, 4287–4298. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [Green Version]
- Roseweir, A.K.; Powell, A.; Horstman, S.L.; Inthagard, J.; Park, J.H.; McMillan, D.C.; Horgan, P.G.; Edwards, J. Src family kinases, HCK and FGR, associate with local inflammation and tumour progression in colorectal cancer. Cell Signal. 2019, 56, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Oneyama, C.; Morii, E.; Okuzaki, D.; Takahashi, Y.; Ikeda, J.; Wakabayashi, N.; Akamatsu, H.; Tsujimoto, M.; Nishida, T.; Aozasa, K.; et al. MicroRNA-mediated upregulation of integrin-linked kinase promotes Src-induced tumor progression. Oncogene 2012, 31, 1623–1635. [Google Scholar] [CrossRef] [Green Version]
- Sancier, F.; Dumont, A.; Sirvent, A.; Paquay de Plater, L.; Edmonds, T.; David, G.; Jan, M.; de Montrion, C.; Coge, F.; Leonce, S.; et al. Specific oncogenic activity of the Src-family tyrosine kinase c-Yes in colon carcinoma cells. PLoS ONE 2011, 6, e17237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, N.M.; Curley, S.A.; Gallick, G.E. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res. 1996, 2, 1397–1404. [Google Scholar] [PubMed]
- Trevino, J.G.; Summy, J.M.; Lesslie, D.P.; Parikh, N.U.; Hong, D.S.; Lee, F.Y.; Donato, N.J.; Abbruzzese, J.L.; Baker, C.H.; Gallick, G.E. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol. 2006, 168, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Gardner-Thorpe, J.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Inhibition of tyrosine kinase Src suppresses pancreatic cancer invasiveness. Surgery 2003, 134, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Barnes, L.A.; Shields, D.J.; Huang, M.; Lau, S.K.; Prevost, N.; Tarin, D.; Shattil, S.J.; Cheresh, D.A. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat. Med. 2009, 15, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, A.; Schenone, S.; Gravina, G.L.; Muzi, P.; Festuccia, C.; Vicentini, C.; Botta, M.; Bologna, M. Pyrazolo[3,4-d]pyrimidines c-Src inhibitors reduce epidermal growth factor-induced migration in prostate cancer cells. Eur. J. Cancer 2006, 42, 2838–2845. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Cai, Y.; Pi, W.; Gao, L.; Shay, C. Augmentation of the anticancer activity of CYT997 in human prostate cancer by inhibiting Src activity. J. Hematol. Oncol. 2017, 10, 118. [Google Scholar] [CrossRef]
- Nam, S.; Kim, D.; Cheng, J.Q.; Zhang, S.; Lee, J.H.; Buettner, R.; Mirosevich, J.; Lee, F.Y.; Jove, R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005, 65, 9185–9189. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Alsaidan, O.A.; Goodwin, O.; Li, Q.; Sulejmani, E.; Han, Z.; Bai, A.; Albers, T.; Beharry, Z.; Zheng, Y.G.; et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res. 2017, 77, 6950–6962. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Siemann, D. c-Src is required for hypoxia-induced metastasis-associated functions in prostate cancer cells. Onco Targets Ther. 2019, 12, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Park, S.I.; Zhang, J.; Phillips, K.A.; Araujo, J.C.; Najjar, A.M.; Volgin, A.Y.; Gelovani, J.G.; Kim, S.J.; Wang, Z.; Gallick, G.E. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008, 68, 3323–3333. [Google Scholar] [CrossRef] [Green Version]
- Mello, A.A.; Leal, M.F.; Rey, J.A.; Pinto, G.R.; Lamarao, L.M.; Montenegro, R.C.; Alves, A.P.; Assumpcao, P.P.; Borges Bdo, N.; Smith, M.C.; et al. Deregulated Expression of SRC, LYN and CKB Kinases by DNA Methylation and Its Potential Role in Gastric Cancer Invasiveness and Metastasis. PLoS ONE 2015, 10, e0140492. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bai, Z.G.; Yin, J.; Wu, G.C.; Zhang, Z.T. Role of c-Src activity in the regulation of gastric cancer cell migration. Oncol. Rep. 2014, 32, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, W.; Okamoto, I.; Yoshida, T.; Okamoto, K.; Takezawa, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Arao, T.; Yanagihara, K.; et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol. Cancer Ther. 2010, 9, 1188–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Yin, S.; Sun, R.; Zhang, S.; Fu, M.; Wu, Y.; Zhang, T.; Khaliq, J.; Li, Y. miR-140-5p suppresses the proliferation, migration and invasion of gastric cancer by regulating YES1. Mol. Cancer 2017, 16, 139. [Google Scholar] [CrossRef]
- Nam, A.R.; Kim, J.W.; Park, J.E.; Bang, J.H.; Jin, M.H.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Im, S.A.; Oh, D.Y.; et al. Src as a Therapeutic Target in Biliary Tract Cancer. Mol. Cancer Ther. 2016, 15, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Pyon, J.K.; Kim, D.W.; Lee, S.H.; Nam, H.S.; Kim, C.H.; Kang, S.G.; Lee, Y.J.; Park, M.Y.; Jeong, D.J.; et al. Elevated c-Src and c-Yes expression in malignant skin cancers. J. Exp. Clin. Cancer Res. 2010, 29, 116. [Google Scholar] [CrossRef] [Green Version]
- Eustace, A.J.; Crown, J.; Clynes, M.; O’Donovan, N. Preclinical evaluation of dasatinib, a potent Src kinase inhibitor, in melanoma cell lines. J. Transl. Med. 2008, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Mesa, T.; Vultur, A.; Lee, F.; Jove, R. Inhibition of Src family kinases with dasatinib blocks migration and invasion of human melanoma cells. Mol. Cancer Res. 2008, 6, 1766–1774. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kim, J.H.; Lee, Y.; Yang, H.; Heo, Y.S.; Bode, A.M.; Lee, K.W.; Dong, Z. Bakuchiol suppresses proliferation of skin cancer cells by directly targeting Hck, Blk, and p38 MAP kinase. Oncotarget 2016, 7, 14616–14627. [Google Scholar] [CrossRef] [Green Version]
- Somlo, G.; Atzori, F.; Strauss, L.C.; Geese, W.J.; Specht, J.M.; Gradishar, W.J.; Rybicki, A.; Sy, O.; Vahdat, L.T.; Cortes, J. Dasatinib plus capecitabine for advanced breast cancer: Safety and efficacy in phase I study CA180004. Clin. Cancer Res. 2013, 19, 1884–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusztai, L.; Moulder, S.; Altan, M.; Kwiatkowski, D.; Valero, V.; Ueno, N.T.; Esteva, F.J.; Avritscher, R.; Qi, Y.; Strauss, L.; et al. Gene signature-guided dasatinib therapy in metastatic breast cancer. Clin. Cancer Res. 2014, 20, 5265–5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Schott, A.F.; Barlow, W.E.; Van Poznak, C.H.; Hayes, D.F.; Moinpour, C.M.; Lew, D.L.; Dy, P.A.; Keller, E.T.; Keller, J.M.; Hortobagyi, G.N. Phase II studies of two different schedules of dasatinib in bone metastasis predominant metastatic breast cancer: SWOG S0622. Breast Cancer Res. Treat. 2016, 159, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.Y.; Wilding, G.; Posadas, E.; Gross, M.; Culine, S.; Massard, C.; Morris, M.J.; Hudes, G.; Calabro, F.; Cheng, S.; et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2009, 15, 7421–7428. [Google Scholar] [CrossRef] [Green Version]
- Mitri, Z.; Nanda, R.; Blackwell, K.; Costelloe, C.M.; Hood, I.; Wei, C.; Brewster, A.M.; Ibrahim, N.K.; Koenig, K.B.; Hortobagyi, G.N.; et al. TBCRC-010: Phase I/II Study of Dasatinib in Combination with Zoledronic Acid for the Treatment of Breast Cancer Bone Metastasis. Clin. Cancer Res. 2016, 22, 5706–5712. [Google Scholar] [CrossRef] [Green Version]
- Twardowski, P.W.; Beumer, J.H.; Chen, C.S.; Kraft, A.S.; Chatta, G.S.; Mitsuhashi, M.; Ye, W.; Christner, S.M.; Lilly, M.B. A phase II trial of dasatinib in patients with metastatic castration-resistant prostate cancer treated previously with chemotherapy. Anticancer Drugs 2013, 24, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.; Reeves, K.; Luo, F.R.; Xu, L.A.; Lee, F.; Clark, E.; Huang, F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: Rationale for patient selection and efficacy monitoring. Genome Biol. 2007, 8, R255. [Google Scholar] [CrossRef] [Green Version]
- Bauman, J.E.; Duvvuri, U.; Gooding, W.E.; Rath, T.J.; Gross, N.D.; Song, J.; Jimeno, A.; Yarbrough, W.G.; Johnson, F.M.; Wang, L.; et al. Randomized, placebo-controlled window trial of EGFR, Src, or combined blockade in head and neck cancer. JCI Insight 2017, 2, e90449. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Ocana, A.; Gil-Martin, M.; Antolin, S.; Atienza, M.; Montano, A.; Ribelles, N.; Urruticoechea, A.; Falcon, A.; Pernas, S.; Orlando, J.; et al. Efficacy and safety of dasatinib with trastuzumab and paclitaxel in first line HER2-positive metastatic breast cancer: Results from the phase II GEICAM/2010-04 study. Breast Cancer Res. Treat. 2019, 174, 693–701. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef]
- Araujo, J.C.; Trudel, G.C.; Saad, F.; Armstrong, A.J.; Yu, E.Y.; Bellmunt, J.; Wilding, G.; McCaffrey, J.; Serrano, S.V.; Matveev, V.B.; et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): A randomised, double-blind phase 3 trial. Lancet Oncol. 2013, 14, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.Y.; Massard, C.; Gross, M.E.; Carducci, M.A.; Culine, S.; Hudes, G.; Posadas, E.M.; Sternberg, C.N.; Wilding, G.; Trudel, G.C.; et al. Once-daily dasatinib: Expansion of phase II study evaluating safety and efficacy of dasatinib in patients with metastatic castration-resistant prostate cancer. Urology 2011, 77, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Herold, C.I.; Chadaram, V.; Peterson, B.L.; Marcom, P.K.; Hopkins, J.; Kimmick, G.G.; Favaro, J.; Hamilton, E.; Welch, R.A.; Bacus, S.; et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin. Cancer Res. 2011, 17, 6061–6070. [Google Scholar] [CrossRef] [Green Version]
- Algazi, A.P.; Weber, J.S.; Andrews, S.C.; Urbas, P.; Munster, P.N.; DeConti, R.C.; Hwang, J.; Sondak, V.K.; Messina, J.L.; McCalmont, T.; et al. Phase I clinical trial of the Src inhibitor dasatinib with dacarbazine in metastatic melanoma. Br. J. Cancer 2012, 106, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Kalinsky, K.; Lee, S.; Rubin, K.M.; Lawrence, D.P.; Iafrarte, A.J.; Borger, D.R.; Margolin, K.A.; Leitao, M.M., Jr.; Tarhini, A.A.; Koon, H.B.; et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 2017, 123, 2688–2697. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dudek, A.Z.; McCann, C.; Ritacco, J.; Southard, N.; Jilaveanu, L.B.; Molinaro, A.; Sznol, M. A phase 2 trial of dasatinib in advanced melanoma. Cancer 2011, 117, 2202–2208. [Google Scholar] [CrossRef]
- Gold, K.A.; Lee, J.J.; Harun, N.; Tang, X.; Price, J.; Kawedia, J.D.; Tran, H.T.; Erasmus, J.J.; Blumenschein, G.R.; William, W.N.; et al. A phase I/II study combining erlotinib and dasatinib for non-small cell lung cancer. Oncologist 2014, 19, 1040–1041. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Jardim, D.L.; Johnson, F.M.; Subbiah, V.; Piha-Paul, S.; Tsimberidou, A.M.; Falchook, G.S.; Karp, D.; Zinner, R.; Wheler, J.; et al. Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Investig. New Drugs 2018, 36, 416–423. [Google Scholar] [CrossRef]
- Hong, D.S.; Choe, J.H.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Piha-Paul, S.; Moulder, S.L.; George, G.C.; Choe, J.M.; Strauss, L.C.; et al. A phase 1 study of gemcitabine combined with dasatinib in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Argiris, A.; Feinstein, T.M.; Wang, L.; Yang, T.; Agrawal, S.; Appleman, L.J.; Stoller, R.G.; Grandis, J.R.; Egloff, A.M. Phase I and pharmacokinetic study of dasatinib and cetuximab in patients with advanced solid malignancies. Investig. New Drugs 2012, 30, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Miyazaki, M.; Okamoto, I.; Ito, Y.; Ueda, K.; Seriu, T.; Nakagawa, K.; Hatake, K. Phase I study of dasatinib (BMS-354825) in Japanese patients with solid tumors. Cancer Sci. 2011, 102, 2058–2064. [Google Scholar] [CrossRef]
- Demetri, G.D.; Lo Russo, P.; MacPherson, I.R.; Wang, D.; Morgan, J.A.; Brunton, V.G.; Paliwal, P.; Agrawal, S.; Voi, M.; Evans, T.R. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 6232–6240. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.J.; Jha, G.; Shoemaker, D.; Herndon, J.E., II; Gu, L.; Barry, W.T.; Crawford, J.; Ready, N. Phase II Study of Dasatinib in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Cancer Invest. 2017, 35, 32–35. [Google Scholar] [CrossRef]
- Johnson, M.L.; Riely, G.J.; Rizvi, N.A.; Azzoli, C.G.; Kris, M.G.; Sima, C.S.; Ginsberg, M.S.; Pao, W.; Miller, V.A. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J. Thorac. Oncol. 2011, 6, 1128–1131. [Google Scholar] [CrossRef] [Green Version]
- Haura, E.B.; Tanvetyanon, T.; Chiappori, A.; Williams, C.; Simon, G.; Antonia, S.; Gray, J.; Litschauer, S.; Tetteh, L.; Neuger, A.; et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Johnson, F.M.; Bekele, B.N.; Feng, L.; Wistuba, I.; Tang, X.M.; Tran, H.T.; Erasmus, J.J.; Hwang, L.L.; Takebe, N.; Blumenschein, G.R.; et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4609–4615. [Google Scholar] [CrossRef] [Green Version]
- Cardin, D.B.; Goff, L.W.; Chan, E.; Whisenant, J.G.; Dan Ayers, G.; Takebe, N.; Arlinghaus, L.R.; Yankeelov, T.E.; Berlin, J.; Merchant, N. Dual Src and EGFR inhibition in combination with gemcitabine in advanced pancreatic cancer: Phase I results: A phase I clinical trial. Investig. New Drugs 2018, 36, 442–450. [Google Scholar] [CrossRef]
- Evans, T.R.J.; Van Cutsem, E.; Moore, M.J.; Bazin, I.S.; Rosemurgy, A.; Bodoky, G.; Deplanque, G.; Harrison, M.; Melichar, B.; Pezet, D.; et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann. Oncol. 2017, 28, 354–361. [Google Scholar] [CrossRef]
- Chee, C.E.; Krishnamurthi, S.; Nock, C.J.; Meropol, N.J.; Gibbons, J.; Fu, P.; Bokar, J.; Teston, L.; O’Brien, T.; Gudena, V.; et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist 2013, 18, 1091–1092. [Google Scholar] [CrossRef] [Green Version]
- Morris, P.G.; Rota, S.; Cadoo, K.; Zamora, S.; Patil, S.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; et al. Phase II Study of Paclitaxel and Dasatinib in Metastatic Breast Cancer. Clin. Breast Cancer 2018, 18, 387–394. [Google Scholar] [CrossRef]
- Mayer, E.L.; Baurain, J.F.; Sparano, J.; Strauss, L.; Campone, M.; Fumoleau, P.; Rugo, H.; Awada, A.; Sy, O.; Llombart-Cussac, A. A phase 2 trial of dasatinib in patients with advanced HER2-positive and/or hormone receptor-positive breast cancer. Clin. Cancer Res. 2011, 17, 6897–6904. [Google Scholar] [CrossRef] [Green Version]
- Fornier, M.N.; Morris, P.G.; Abbruzzi, A.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; Modi, S.; Seidman, A.D.; et al. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann. Oncol. 2011, 22, 2575–2581. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Thomas, D.G.; von Mehren, M.; Patel, S.; Samuels, B.; Choy, E.; D’Amato, G.; Staddon, A.P.; Ganjoo, K.N.; et al. Association of Dasatinib With Progression-Free Survival Among Patients With Advanced Gastrointestinal Stromal Tumors Resistant to Imatinib. JAMA Oncol. 2018, 4, 814–820. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Keohan, M.L.; Dickson, M.A.; Gounder, M.M.; Chi, P.; Loo, J.K.; Gaffney, L.; Schneider, L.; Patel, Z.; et al. Combined KIT and CTLA-4 Blockade in Patients with Refractory GIST and Other Advanced Sarcomas: A Phase Ib Study of Dasatinib plus Ipilimumab. Clin. Cancer Res. 2017, 23, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Schuetze, S.M.; Wathen, J.K.; Lucas, D.R.; Choy, E.; Samuels, B.L.; Staddon, A.P.; Ganjoo, K.N.; von Mehren, M.; Chow, W.A.; Loeb, D.M.; et al. SARC009: Phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer 2016, 122, 868–874. [Google Scholar] [CrossRef]
- Secord, A.A.; Teoh, D.K.; Barry, W.T.; Yu, M.; Broadwater, G.; Havrilesky, L.J.; Lee, P.S.; Berchuck, A.; Lancaster, J.; Wenham, R.M. A phase I trial of dasatinib, an SRC-family kinase inhibitor, in combination with paclitaxel and carboplatin in patients with advanced or recurrent ovarian cancer. Clin. Cancer Res. 2012, 18, 5489–5498. [Google Scholar] [CrossRef] [Green Version]
- Parseghian, C.M.; Parikh, N.U.; Wu, J.Y.; Jiang, Z.Q.; Henderson, L.; Tian, F.; Pastor, B.; Ychou, M.; Raghav, K.; Dasari, A.; et al. Dual Inhibition of EGFR and c-Src by Cetuximab and Dasatinib Combined with FOLFOX Chemotherapy in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 4146–4154. [Google Scholar] [CrossRef] [Green Version]
- Strickler, J.H.; McCall, S.; Nixon, A.B.; Brady, J.C.; Pang, H.; Rushing, C.; Cohn, A.; Starodub, A.; Arrowood, C.; Haley, S.; et al. Phase I study of dasatinib in combination with capecitabine, oxaliplatin and bevacizumab followed by an expanded cohort in previously untreated metastatic colorectal cancer. Investig. New Drugs 2014, 32, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.M.; Costa, D.B.; Heist, R.S.; Garcia, E.; Lindeman, N.I.; Sholl, L.M.; Oxnard, G.R.; Johnson, B.E.; Hammerman, P.S. Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J. Thorac. Oncol. 2013, 8, 1434–1437. [Google Scholar] [CrossRef] [Green Version]
- Stabile, L.P.; Egloff, A.M.; Gibson, M.K.; Gooding, W.E.; Ohr, J.; Zhou, P.; Rothenberger, N.J.; Wang, L.; Geiger, J.L.; Flaherty, J.T.; et al. IL6 is associated with response to dasatinib and cetuximab: Phase II clinical trial with mechanistic correlatives in cetuximab-resistant head and neck cancer. Oral Oncol. 2017, 69, 38–45. [Google Scholar] [CrossRef]
- Brooks, H.D.; Glisson, B.S.; Bekele, B.N.; Johnson, F.M.; Ginsberg, L.E.; El-Naggar, A.; Culotta, K.S.; Takebe, N.; Wright, J.; Tran, H.T.; et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer 2011, 117, 2112–2119. [Google Scholar] [CrossRef]
- Lara, P.N., Jr.; Longmate, J.; Evans, C.P.; Quinn, D.I.; Twardowski, P.; Chatta, G.; Posadas, E.; Stadler, W.; Gandara, D.R. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: A California Cancer Consortium study. Anticancer Drugs 2009, 20, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, Y.; Onozawa, Y.; Kurata, T.; Yasui, H.; Goto, I.; Yamazaki, K.; Machida, N.; Watanabe, J.; Shimada, H.; Shi, X.; et al. First report of the safety, tolerability, and pharmacokinetics of the Src kinase inhibitor saracatinib (AZD0530) in Japanese patients with advanced solid tumours. Investig. New Drugs 2013, 31, 108–114. [Google Scholar] [CrossRef]
- Gucalp, A.; Sparano, J.A.; Caravelli, J.; Santamauro, J.; Patil, S.; Abbruzzi, A.; Pellegrino, C.; Bromberg, J.; Dang, C.; Theodoulou, M.; et al. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin. Breast Cancer 2011, 11, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Laurie, S.A.; Goss, G.D.; Shepherd, F.A.; Reaume, M.N.; Nicholas, G.; Philip, L.; Wang, L.; Schwock, J.; Hirsh, V.; Oza, A.; et al. A phase II trial of saracatinib, an inhibitor of src kinases, in previously-treated advanced non-small-cell lung cancer: The princess margaret hospital phase II consortium. Clin. Lung Cancer 2014, 15, 52–57. [Google Scholar] [CrossRef]
- Hannon, R.A.; Finkelman, R.D.; Clack, G.; Iacona, R.B.; Rimmer, M.; Gossiel, F.; Baselga, J.; Eastell, R. Effects of Src kinase inhibition by saracatinib (AZD0530) on bone turnover in advanced malignancy in a Phase I study. Bone 2012, 50, 885–892. [Google Scholar] [CrossRef]
- Posadas, E.M.; Ahmed, R.S.; Karrison, T.; Szmulewitz, R.Z.; O’Donnell, P.H.; Wade, J.L., III; Shen, J.; Gururajan, M.; Sievert, M.; Stadler, W.M. Saracatinib as a metastasis inhibitor in metastatic castration-resistant prostate cancer: A University of Chicago Phase 2 Consortium and DOD/PCF Prostate Cancer Clinical Trials Consortium Study. Prostate 2016, 76, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Renouf, D.J.; Moore, M.J.; Hedley, D.; Gill, S.; Jonker, D.; Chen, E.; Walde, D.; Goel, R.; Southwood, B.; Gauthier, I.; et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2012, 30, 779–786. [Google Scholar] [CrossRef]
- Kaye, S.; Aamdal, S.; Jones, R.; Freyer, G.; Pujade-Lauraine, E.; de Vries, E.G.; Barriuso, J.; Sandhu, S.; Tan, D.S.; Hartog, V.; et al. Phase I study of saracatinib (AZD0530) in combination with paclitaxel and/or carboplatin in patients with solid tumours. Br. J. Cancer 2012, 106, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Trarbach, T.; Schultheis, B.; Gauler, T.C.; Schneider, V.; Strumberg, D.; Eberhardt, W.E.; Le Scouiller, S.; Marotti, M.; Brown, K.H.; Drevs, J. Phase I open-label study of cediranib, an oral inhibitor of VEGF signalling, in combination with the oral Src inhibitor saracatinib in patients with advanced solid tumours. Investig. New Drugs 2012, 30, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Brown, J.; Larkin, J.; Jones, R.; Ralph, C.; Hawkins, R.; Chowdhury, S.; Boleti, E.; Bhal, A.; Fife, K.; et al. A randomized, double-blind phase II study evaluating cediranib versus cediranib and saracatinib in patients with relapsed metastatic clear-cell renal cancer (COSAK). Ann. Oncol. 2016, 27, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Gubens, M.A.; Burns, M.; Perkins, S.M.; Pedro-Salcedo, M.S.; Althouse, S.K.; Loehrer, P.J.; Wakelee, H.A. A phase II study of saracatinib (AZD0530), a Src inhibitor, administered orally daily to patients with advanced thymic malignancies. Lung Cancer 2015, 89, 57–60. [Google Scholar] [CrossRef]
- Mackay, H.J.; Au, H.J.; McWhirter, E.; Alcindor, T.; Jarvi, A.; MacAlpine, K.; Wang, L.; Wright, J.J.; Oza, A.M. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: A trial of the PMH phase II consortium. Investig. New Drugs 2012, 30, 1158–1163. [Google Scholar] [CrossRef]
- Fury, M.G.; Baxi, S.; Shen, R.; Kelly, K.W.; Lipson, B.L.; Carlson, D.; Stambuk, H.; Haque, S.; Pfister, D.G. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011, 31, 249–253. [Google Scholar]
- Gangadhar, T.C.; Clark, J.I.; Karrison, T.; Gajewski, T.F. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Investig. New drugs 2013, 31, 769–773. [Google Scholar] [CrossRef]
- Daud, A.I.; Krishnamurthi, S.S.; Saleh, M.N.; Gitlitz, B.J.; Borad, M.J.; Gold, P.J.; Chiorean, E.G.; Springett, G.M.; Abbas, R.; Agarwal, S.; et al. Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Campone, M.; Bondarenko, I.; Brincat, S.; Hotko, Y.; Munster, P.N.; Chmielowska, E.; Fumoleau, P.; Ward, R.; Bardy-Bouxin, N.; Leip, E.; et al. Phase II study of single-agent bosutinib, a Src/Abl tyrosine kinase inhibitor, in patients with locally advanced or metastatic breast cancer pretreated with chemotherapy. Ann. Oncol. 2012, 23, 610–617. [Google Scholar] [CrossRef]
- Messersmith, W.A.; Krishnamurthi, S.S.; Hewes, B.A.; Zacharchuk, C.M.; Abbas, R.; Martins, P.; Dowling, E.; Volkert, A.; Martin, E.; Daud, A.I. Bosutinib (SKI-606), a dual Src/Abl tyrosine kinase inhibitor: Preliminary results from a phase 1 study in patients with advanced malignant solid tumors. J. Clin. Oncol. 2007, 25, 3552. [Google Scholar] [CrossRef]
- Moy, B.; Neven, P.; Lebrun, F.; Bellet, M.; Xu, B.; Sarosiek, T.; Chow, L.; Goss, P.; Zacharchuk, C.; Leip, E.; et al. Bosutinib in combination with the aromatase inhibitor letrozole: A phase II trial in postmenopausal women evaluating first-line endocrine therapy in locally advanced or metastatic hormone receptor-positive/HER2-negative breast cancer. Oncologist 2014, 19, 348–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moy, B.; Neven, P.; Lebrun, F.; Bellet, M.; Xu, B.; Sarosiek, T.; Chow, L.; Goss, P.; Zacharchuk, C.; Leip, E.; et al. Bosutinib in combination with the aromatase inhibitor exemestane: A phase II trial in postmenopausal women with previously treated locally advanced or metastatic hormone receptor-positive/HER2-negative breast cancer. Oncologist 2014, 19, 346–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakoff, S.J.; Wang, D.; Campone, M.; Calles, A.; Leip, E.; Turnbull, K.; Bardy-Bouxin, N.; Duvillie, L.; Calvo, E. Bosutinib plus capecitabine for selected advanced solid tumours: Results of a phase 1 dose-escalation study. Br. J. Cancer 2014, 111, 2058–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodcock, V.K.; Clive, S.; Wilson, R.H.; Coyle, V.M.; Stratford, M.R.L.; Folkes, L.K.; Eastell, R.; Barton, C.; Jones, P.; Kazmi-Stokes, S.; et al. A first-in-human phase I study to determine the maximum tolerated dose of the oral Src/ABL inhibitor AZD0424. Br. J. Cancer 2018, 118, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Protein Kinase | Tissue Distribution | Solid Tumor Distribution | |
|---|---|---|---|
| Level of Expression | Tumor (*) | ||
| Src | most | strong | cervical, head and neck, pancreatic, skin, urothelial |
| moderate | colorectal, lung, stomach | ||
| weak | carcinoid, cervical | ||
| Yes | most | moderate | most (>60% = breast, colorectal, head and neck, liver, ovarian, prostate, testis, thyroid, urothelial) |
| Frk | most | strong | thyroid |
| weak to moderate | carcinoid, colorectal, endometrial, liver, melanoma, renal, urothelial | ||
| Lyn | most | moderate to strong | liver, stomach |
| weak | carcinoid, head and neck, thyroid | ||
| Fyn | brain, endocrine tissues, female tissues, hematopoietic cells, liver | moderate | glioma |
| weak | carcinoid, thyroid | ||
| Blk | hematopoietic cells, lung | moderate to strong | endometrial |
| Fgr | hematopoietic cells, lung | weak | Carcinoid, colorectal, renal, thyroid |
| Hck | hematopoietic cells, lung | strong | endometrial, lung, renal, stomach |
| weak | carcinoid, glioma, liver, ovarian, pancreas, skin | ||
| Lck | hematopoietic cells | negative | none |
| Srms | gastrointestinal, male tissue (**) | strong | colorectal, ovarian, prostate |
| moderate | most | ||
| Cancer | Phase | Combination | Efficacy | Dose | Year of Publication | Ref. |
|---|---|---|---|---|---|---|
| Advanced solid tumors | I | crizotinib | PR = 2% SD = 5% (mainly sarcoma and prostate cancer) | 140 mg daily | 2018 | [130] |
| I | gemcitabine | PR = 25% (pancreatic cancer) | From 70 mg daily to 100 mg daily | 2012 | [131] | |
| I | cetuximab | SD = 43% (better PFS in patients with low baseline TGF-α levels) | 100 mg, 150 mg, 200 mg daily | 2012 | [132] | |
| I | none | SD = 19% | 100 mg, 150 mg, 200 mg daily | 2011 | [133] | |
| I | none | SD = 16% | 35 to 160 mg twice daily | 2009 | [134] | |
| Advanced Non-Small Cell Lung Cancer | II | none | No response | 70 mg twice daily | 2017 | [135] |
| I/II | erlotinib | PR = 15% (EGFR mutated population) | 70 mg daily | 2014 | [129] | |
| II | erlotinib | PR = 0% (EGFR-mutant and acquired resistance to EGFR-Tirosine Kinase Inhibitors) | 70 mg twice daily, 100 mg daily | 2011 | [136] | |
| I/II | erlotinib | PR = 7% | 50 mg twice, or 70 mg twice daily, or 140 mg daily | 2010 | [137] | |
| II | none | SD = 21% | 100 mg twice daily reduced to 100 mg + 50 mg daily | 2010 | [138] | |
| Advanced pancreatic cancer | I | Erlotinib gemcitabine | SD = 69% | 70 mg daily reduced to 50 mg daily | 2018 | [139] |
| II | gemcitabine | No response | 100 mg daily | 2017 | [140] | |
| II | none | SD = 29% Metastatic cases | 100 mg twice daily reduced to 70 mg twice daily | 2013 | [141] | |
| Advanced breast cancer | II | trastuzumab paclitaxel | PR = 69% SD = 10% (HER2 + metastatic cases) | 100 mg daily | 2019 | [121] |
| II | paclitaxel | CR = 3% PR = 20% (mainly ER+) | 120 mg daily | 2018 | [142] | |
| I/II | zolendronic acid | CR + PR = 23% SD = 13% | From 70 mg twice daily to 100 mg daily | 2016 | [116] | |
| II | none | PR = 4% (bone metastases) | 70 mg twice daily and 100 mg daily | 2016 | [114] | |
| I | capecitabine | PR = 24% SD = 32% | 50 or 70 mg twice or 100 mg daily | 2013 | [111] | |
| II | none | PR = 4% PR = 100% (HER2 + ER+) | 100 mg twice daily reduced to 70 mg twice daily | 2011 | [143] | |
| I | paclitaxel | PR = 31% SD = 29% | 70–120 mg daily | 2011 | [144] | |
| Imatinib-refractory metastatic Gastrointestinal Stromal Tumors | II | none | PR = 25% (PFS = 50% in pSRC+) | 70 mg twice daily | 2018 | [145] |
| High-grade and refractory Advanced Sarcoma | Ib | ipilimumab | No response | from 70mg to 100 mg or 70 mg twice, daily | 2017 | [146] |
| II | none | SD (6 months) = 10–12% (leiomyosarcoma, osteosarcoma, undifferentiated pleomorphic sarcoma) | 100 mg daily | 2016 | [147] | |
| Advanced ovarian cancer | I | paclitaxel carboplatin | PR = 25% SD = 50% | 100, 120, 150 mg daily | 2012 | [148] |
| Metastatic colorectal cancer | Ib/II | FOLFOX chemiotherapy w and w/o cetuximab | SD = 47% PR = 20% | 100, 150, 200 mg daily | 2017 | [149] |
| I | capecitabine oxaliplatin bevacizumab | PR = 10% SD = 60% | 50 mg twice or 70 mg daily | 2013 | [150] | |
| Metastatic castration resistant prostate cancer | II | none | SD = 19% PR = 4% | 70 mg twice reduced to 100 mg daily | 2013 | [117] |
| III | docetaxel | No response | 100 mg daily | 2013 | [123] | |
| II | none | SD (3 months) = 44% SD (6 months) = 17% | 100 mg daily | 2011 | [124] | |
| II | none | SD (3 months) = 43% SD (6 months) = 19% (Mainly bone metastases) | 100 mg twice daily and 50 mg twice daily | 2009 | [115] | |
| Advanced melanoma | II | none | PR = 6% PR (in KIT-) = 100% (stage IV, Mucosal, acral, or vulvovaginal) | 70 mg twice daily | 2017 | [127] |
| I | dacarbazine | PR+SD = 62% (metastatic cases) | 70 mg twice daily | 2012 | [126] | |
| II | none | PR = 6% (no response in patients with mutated KIT) | 100 mg twice daily reduced to 70 mg twice daily | 2011 | [128] | |
| Advanced squamous cell carcinoma | II | none | No response Intolerable toxicity in the majority of cases. | 140 mg daily | 2013 | [151] |
| Metastatic Head and Neck Squamous Cell Carcinoma | II | cetuximab | SD = 36% (mainly in low serum IL6) | 150 mg daily | 2017 | [152] |
| I | with or without erlotinib | No benefit respect to erlotinib alone (PR in low pStat3) | 100 mg daily | 2017 | [119] | |
| II | none | SD = 17% | 100 mg twice daily reduced to 150 mg daily or 50 mg twice daily | 2011 | [153] |
| Cancer | Phase | Combination | Efficacy | Dose | Year of Publication | Ref. |
|---|---|---|---|---|---|---|
| Advanced solid tumors | I | none | SD = 25% | 50, 125, 175 mg daily | 2013 | [155] |
| I | cedinarib | SD = 63% | 175 mg daily | 2012 | [162] | |
| I | paclitaxel (PTX) and/or carboplatin (CBP) | PR = 11% (PTX+CBP) PR = 21% (PTX) SD ≤ 15% | 125, 175, 225, 250, 300 mg daily | 2012 | [161] | |
| Advanced pancreatic cancer | I/II | gemcitabine | SD (4 months) = 23% PR = 9% | 175 mg daily | 2012 | [160] |
| Advanced NSCLC | II | none | PR = 5% SD = 11% | 175 mg daily | 2014 | [157] |
| Advanced thymic tumors | II | none | SD = 43% | 175 mg daily | 2015 | [164] |
| Advanced gastric or gastro Oesophageal Junction (GEJ) Adenocarcinoma | II | none | SD = 18% | 175 mg daily | 2012 | [165] |
| Advanced CR prostate cancer | II | none | SD (2 months) = 26% | 175 mg daily | 2016 | [159] |
| II | none | SD (5 months) = 11% | 175 mg daily | 2009 | [154] | |
| Advanced breast cancer | II | none | No response (ER-metastatic cases) | 175 mg daily | 2011 | [156] |
| Recurrent and metastatic HNSCC | II | none | No response | 175 mg daily | 2011 | [166] |
| Metastatic Clear-Cell Renal Cancer | II | cediranib | PR = 15% | 175 mg daily | 2016 | [163] |
| Metastatic melanoma | II | none | SD = 9% | 175 mg daily | 2013 | [167] |
| Relapsed ovarian cancer | III | paclitaxel | SD = 42% vs. 97% (placebo) | 175 mg daily | 2015 | ClinicalTrials.gov Identifier: NCT01196741 |
| Cancer | Phase | Combination | Efficacy | Dose | Year | Ref. |
|---|---|---|---|---|---|---|
| Advanced solid tumors | I | capecitabine | SD = 64% (colorectal cancer) SD = 45% (breast cancer) | 300 mg daily | 2014 | [173] |
| I | none | SD = 47% (NSCLC) SD = 29% (colorectal) SD = 22% (pancreas cancer) | Escalating from 600 mg daily | 2012 | [168] | |
| I | none | SD = 12% | 50 to 600 mg daily | 2007 | [170] | |
| Advanced breast cancer | II | exemestane | PR = 2% SD = 7% (ER+ HER2-) | 400 mg or 300 mg daily | 2014 | [172] |
| II | letrozole | PR = 6% SD = 6% (ER+HER2-) | 400 mg daily | 2014 | [171] | |
| II | none | PR = 5.5% SD = 32.9% SD = 41.7% (HER2+) | 400 mg daily | 2012 | [169] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far. Cancers 2020, 12, 1448. https://doi.org/10.3390/cancers12061448
Martellucci S, Clementi L, Sabetta S, Mattei V, Botta L, Angelucci A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far. Cancers. 2020; 12(6):1448. https://doi.org/10.3390/cancers12061448
Chicago/Turabian StyleMartellucci, Stefano, Letizia Clementi, Samantha Sabetta, Vincenzo Mattei, Lorenzo Botta, and Adriano Angelucci. 2020. "Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far" Cancers 12, no. 6: 1448. https://doi.org/10.3390/cancers12061448
APA StyleMartellucci, S., Clementi, L., Sabetta, S., Mattei, V., Botta, L., & Angelucci, A. (2020). Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far. Cancers, 12(6), 1448. https://doi.org/10.3390/cancers12061448