SPOP Deregulation Improves the Radiation Response of Prostate Cancer Models by Impairing DNA Damage Repair

 , , ,
, , ,  , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
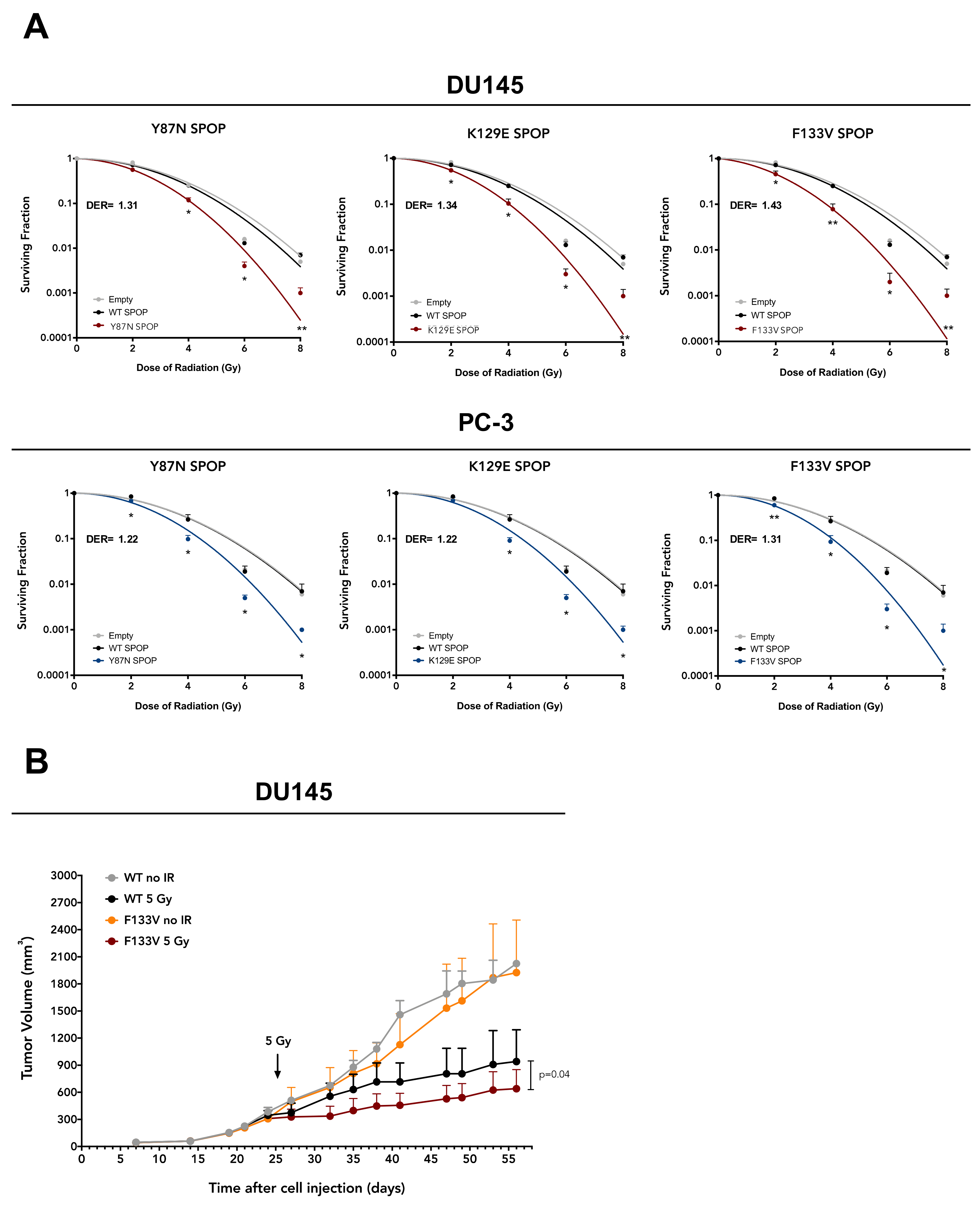
2.1. SPOP Mutation Sensitizes PCa Models to Ionizing Radiation
2.2. SPOP Mutation Affects PCa Cell Radiation Response by Impairing Homologous Recombination
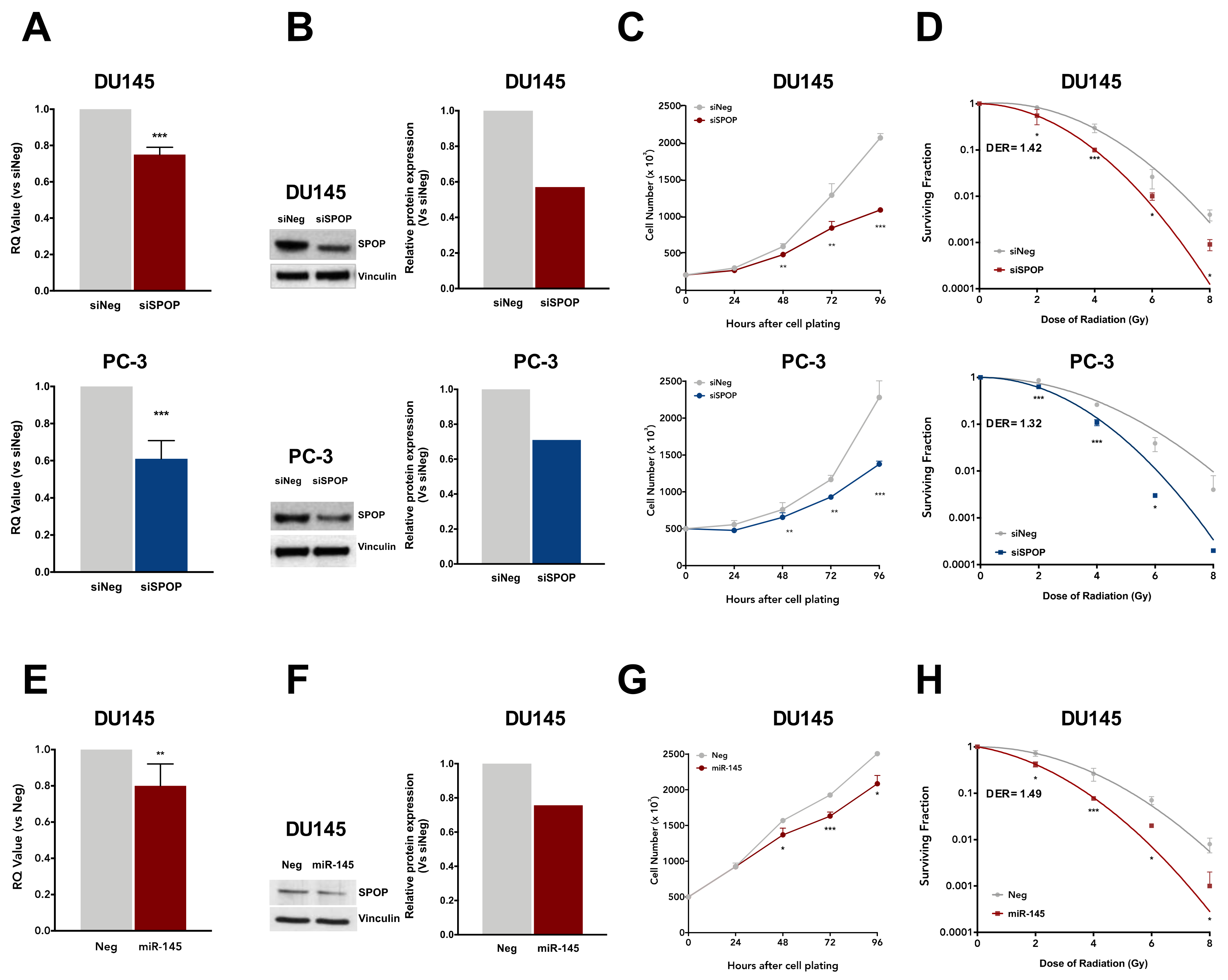
2.3. Knockdown of SPOP Enhances PCa Cell Response to Ionizing Radiation
2.4. SPOP Knockdown Impairs Homologous Recombination via RAD51 and CHK1 Downregulation
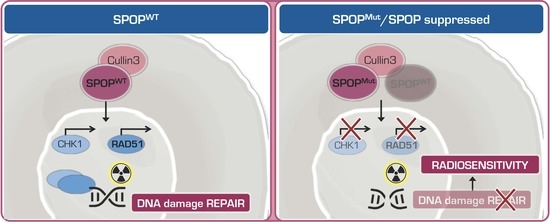
3. Discussion
4. Materials and Methods
4.1. Experimental Models
4.2. Cell Transfection
4.3. Cell Proliferation
4.4. Clonogenic Assay
4.5. In Vivo Experiments
4.6. Total RNA Extraction and RT-qPCR
4.7. Immunoblotting Analyses
4.8. Immunofluorescence
4.9. Bioinformatic Analyses
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-Substrate Complexes: Insights into Molecular Architectures of BTB-Cul3 Ubiquitin Ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Ci, W.; Karmakar, S.; Chen, K.; Dhar, R.; Fan, Z.; Guo, Z.; Zhang, J.; Ke, Y.; Wang, L.; et al. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell 2014, 25, 455–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. eLife 2015, 4, e09207. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Theurillat, J.P.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptoristhe keytranscriptional mediatorofthe tumor suppressor SPOP in prostate cancer. Cancer Res. 2014, 74, 5631–5643. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of Full-Length Androgen Receptor by Wild-Type SPOP, but Not Prostate-Cancer-Associated Mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Hjorth-Jensen, K.; Maya-Mendoza, A.; Dalgaard, N.; Sigurðsson, J.O.; Bartek, J.; Iglesias-Gato, D.; Olsen, J.V.; Flores-Morales, A. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res. 2018, 46, 9484–9495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, H.; Sun, M.; Yang, J.; Zhang, W.; Han, S.; Xu, B. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis 2014, 35, 1691–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Zhang, D.; Cai, M.; Luo, Z.; Zhu, Y.; Gong, L.; Lei, Y.; Tan, X.; Zhu, Q.; Han, S. SPOP regulates the DNA damage response and lung adenocarcinoma cell response to radiation. Am. J. Cancer Res. 2019, 9, 1469. [Google Scholar]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef]
- Tetreault-Laflamme, A.; Crook, J. Options for Salvage of Radiation Failures for Prostate Cancer. Semin. Radiat. Oncol. 2017, 27, 67–78. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Emerging roles of radioresistance in prostate cancer metastasis and radiation therapy. Cancer Metastasis Rev. 2014, 33, 469–496. [Google Scholar] [CrossRef]
- Huang, C.J.; Chen, H.Y.; Lin, W.Y.; Choo, K.B. Differential expression of speckled POZ protein, SPOP: Putative regulation by miR-145. J. Biosci. 2014, 39, 401–413. [Google Scholar] [CrossRef]
- Gong, P.; Zhang, T.; He, D.; Hsieh, J.-T. MicroRNA-145 Modulates Tumor Sensitivity to Radiation in Prostate Cancer. Radiat. Res. 2015, 184, 630–638. [Google Scholar] [CrossRef] [PubMed]
- El Bezawy, R.; Cominetti, D.; Fenderico, N.; Zuco, V.; Beretta, G.L.; Dugo, M.; Arrighetti, N.; Stucchi, C.; Rancati, T.; Valdagni, R.; et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer Lett. 2017, 395, 53–62. [Google Scholar] [CrossRef] [PubMed]
- El Bezawy, R.; Tinelli, S.; Tortoreto, M.; Doldi, V.; Zuco, V.; Folini, M.; Stucchi, C.; Rancati, T.; Valdagni, R.; Gandellini, P.; et al. miR-205 enhances radiation sensitivity of prostate cancer cells by impairing DNA damage repair through PKCε and ZEB1 inhibition. J. Exp. Clin. Cancer Res. 2019, 38, 51. [Google Scholar] [CrossRef] [PubMed]
- Bodgi, L.; Canet, A.; Pujo-Menjouet, L.; Lesne, A.; Victor, J.M.; Foray, N. Mathematical models of radiation action on living cells: From the target theory to the modern approaches. A historical and critical review. J. Theor. Biol. 2016, 394, 93–101. [Google Scholar] [CrossRef]
- Citrin, D.E.; Shankavaram, U.; Horton, J.A.; Shield, W.; Zhao, S.; Asano, H.; White, A.; Sowers, A.; Thetford, A.; Chung, E.J. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J. Natl. Cancer Inst. 2013, 105, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Seol, M.A.; Jung, U.; Eom, H.S.; Kim, S.H.; Park, H.R.; Jo, S.K. Prolonged expression of senescence markers in mice exposed to gamma-irradiation. J. Vet. Sci. 2012, 13, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Hatano, K.; Kumar, B.; Zhang, Y.; Coulter, J.B.; Hedayati, M.; Mears, B.; Ni, X.; Kudrolli, T.A.; Chowdhury, W.H.; Rodriguez, R.; et al. A functional screen identifies miRNAs that inhibit DNA repair and sensitize prostate cancer cells to ionizing radiation. Nucleic Acids Res. 2015, 43, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.T.; Liu, J.; Tang, K.; Wong, Y.C.; Khanna, K.K.; Ling, M.T. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS ONE 2012, 7, e51108. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.S.; Baliga, N.T.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Bezawy, R.; Tripari, M.; Percio, S.; Cicchetti, A.; Tortoreto, M.; Stucchi, C.; Tinelli, S.; Zuco, V.; Doldi, V.; Gandellini, P.; et al. SPOP Deregulation Improves the Radiation Response of Prostate Cancer Models by Impairing DNA Damage Repair. Cancers 2020, 12, 1462. https://doi.org/10.3390/cancers12061462
El Bezawy R, Tripari M, Percio S, Cicchetti A, Tortoreto M, Stucchi C, Tinelli S, Zuco V, Doldi V, Gandellini P, et al. SPOP Deregulation Improves the Radiation Response of Prostate Cancer Models by Impairing DNA Damage Repair. Cancers. 2020; 12(6):1462. https://doi.org/10.3390/cancers12061462
Chicago/Turabian StyleEl Bezawy, Rihan, Martina Tripari, Stefano Percio, Alessandro Cicchetti, Monica Tortoreto, Claudio Stucchi, Stella Tinelli, Valentina Zuco, Valentina Doldi, Paolo Gandellini, and et al. 2020. "SPOP Deregulation Improves the Radiation Response of Prostate Cancer Models by Impairing DNA Damage Repair" Cancers 12, no. 6: 1462. https://doi.org/10.3390/cancers12061462
APA StyleEl Bezawy, R., Tripari, M., Percio, S., Cicchetti, A., Tortoreto, M., Stucchi, C., Tinelli, S., Zuco, V., Doldi, V., Gandellini, P., Valdagni, R., & Zaffaroni, N. (2020). SPOP Deregulation Improves the Radiation Response of Prostate Cancer Models by Impairing DNA Damage Repair. Cancers, 12(6), 1462. https://doi.org/10.3390/cancers12061462