Development of Folate Receptor−Targeted PET Radiopharmaceuticals for Tumor Imaging—A Bench-to-Bedside Journey

Abstract
:1. Introduction
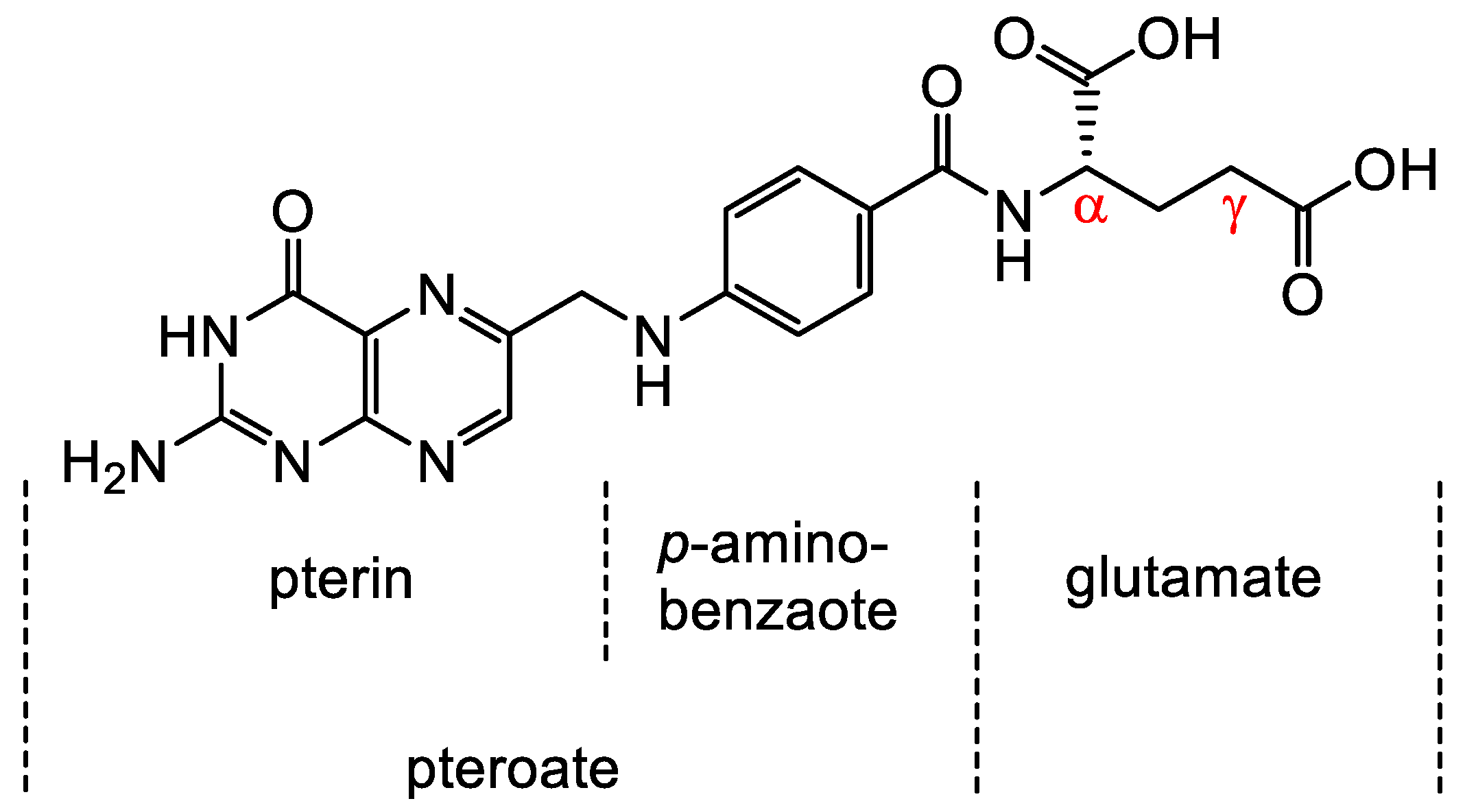
The Family of Folates and the Folate Receptor
2. Structure-Activity-Relationship and Library Design of Radiolabeled Folate Derivatives
2.1. Chemical Modifications of Folic Acid to Produce Radiolabeled Derivatives
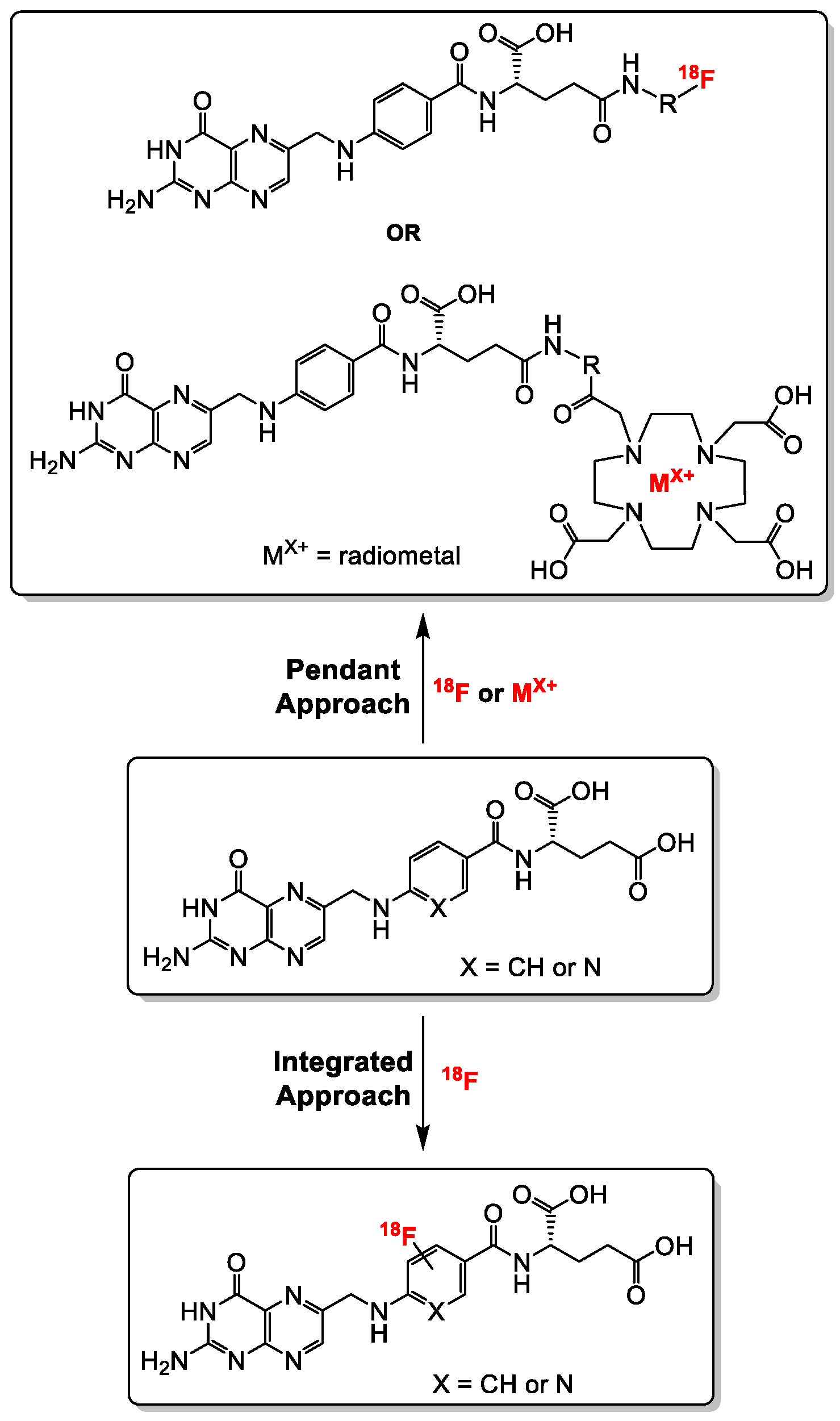
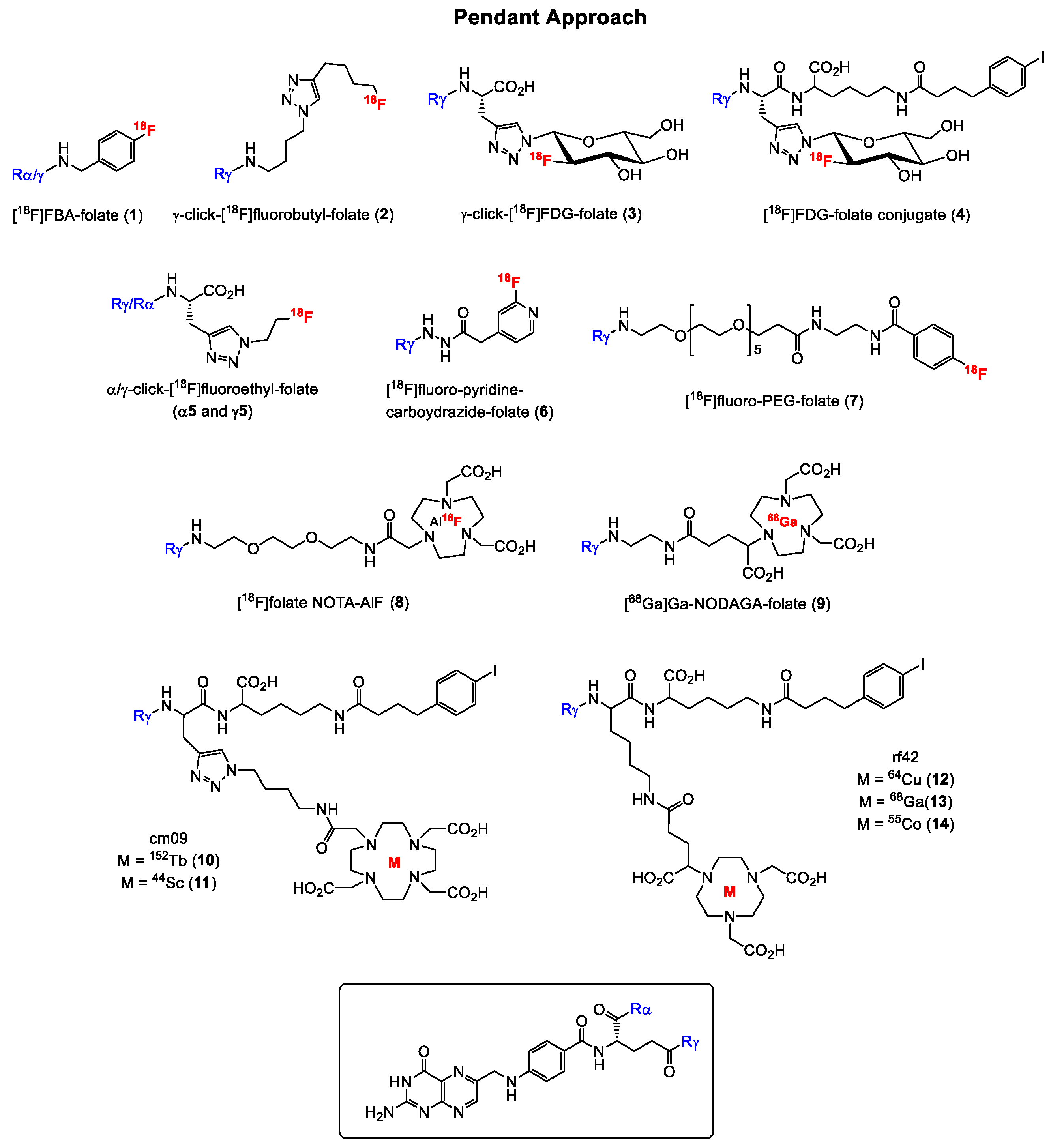
2.1.1. α-and γ-Derivatization of the Glutamate Groups in Folic Acid—The Pendant Approach
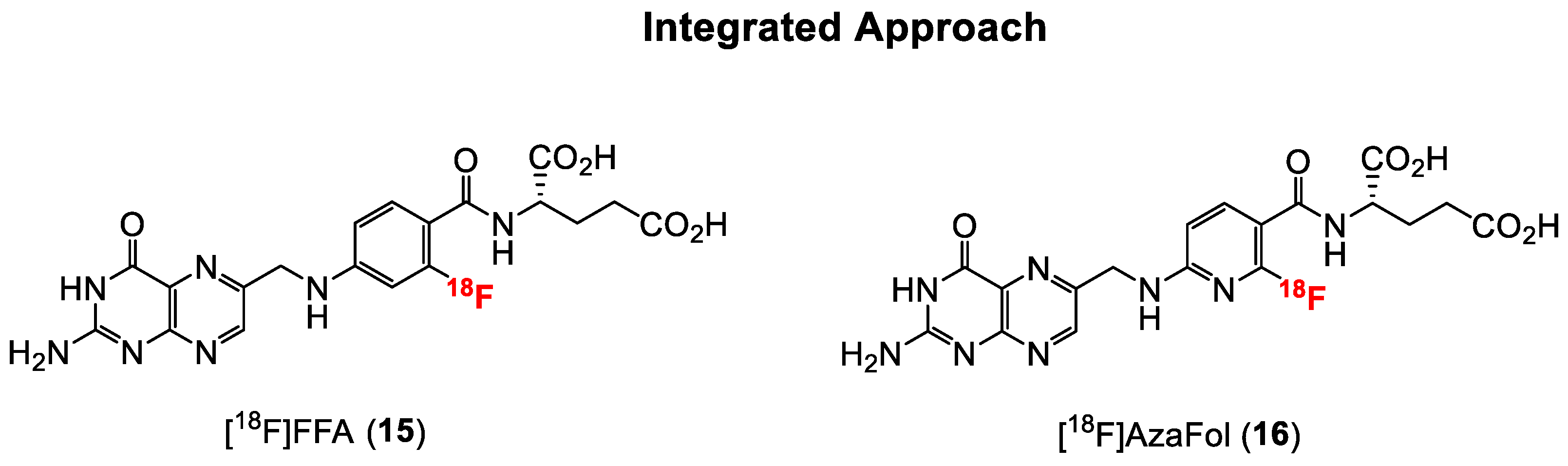
2.1.2. Direct Labeling of Folate Backbone—The Integrated Approach
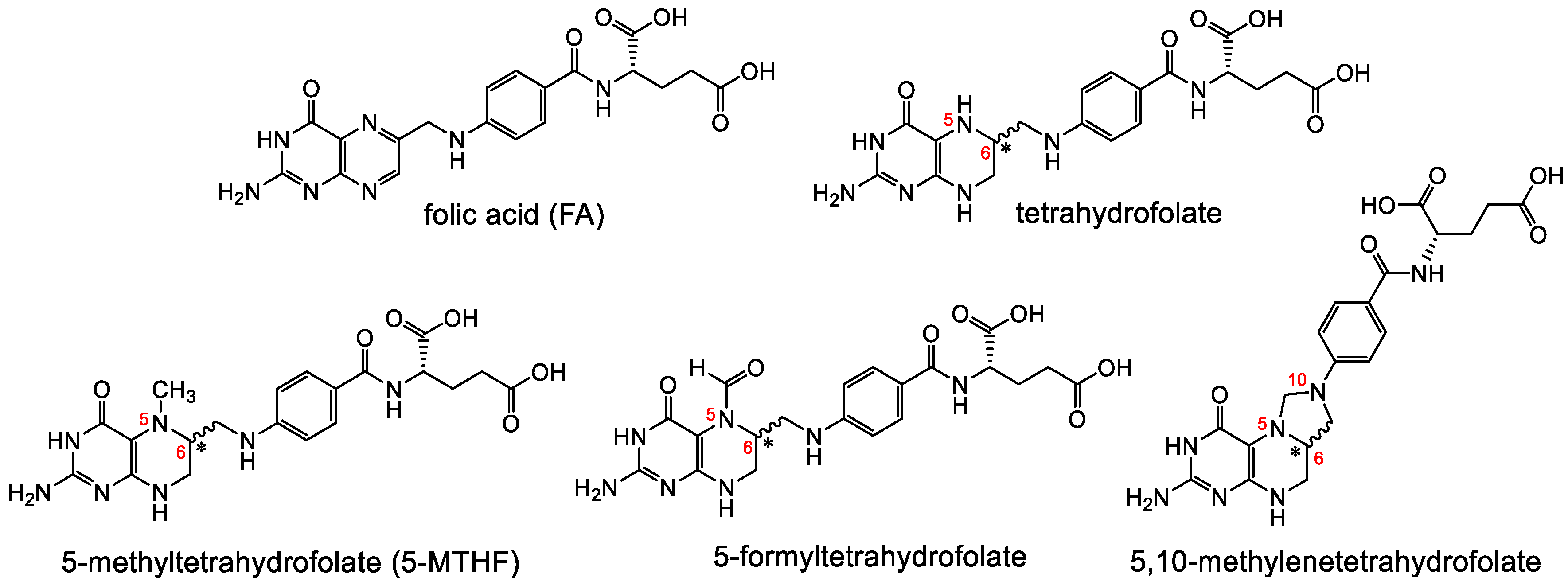
2.2. Reduced Folate Derivatives
3. Selecting Lead Candidates for Further Investigation Leading to Clinical Studies
4. Important Considerations for Translating Folate Radiopharmaceuticals for Human Studies
5. Conclusions
Funding
Conflicts of Interest
References
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Sirotnak, F.M.; Tolner, B. Carrier-mediated membrane transport of folates in mammalian cells. Annu. Rev. Nutr. 1999, 19, 91–122. [Google Scholar] [CrossRef] [PubMed]
- Desmoulin, S.K.; Hou, Z.; Gangjee, A.; Matherly, L.H. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol. Ther. 2012, 13, 1355–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, A.C. Folate Receptors. Annu. Rev. Nutr. 1996, 16, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Spiegelstein, O.; Eudy, J.D.; Finnell, R.H. Identification of two putative novel folate receptor genes in humans and mouse. Gene 2000, 258, 117–125. [Google Scholar] [CrossRef]
- Whetstine, J.R.; Flatley, R.M.; Matherly, L.H. The human reduced folate carrier gene is ubiquitously and differentially expressed in normal human tissues: Identification of seven non-coding exons and characterization of a novel promoter. Biochem. J. 2002, 367, 629–640. [Google Scholar] [CrossRef]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef] [Green Version]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Weitman, S.D.; Lark, R.H.; Coney, L.R.; Fort, D.W.; Frasca, V.; Zurawski, V.R.; Kamen, B.A. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992, 52, 3396–3401. [Google Scholar] [PubMed]
- Weitman, S.D.; Weinberg, A.G.; Coney, L.R.; Zurawski, V.R.; Jennings, D.S.; Kamen, B.A. Cellular localization of the folate receptor: Potential role in drug toxicity and folate homeostasis. Cancer Res. 1992, 52, 6708–6711. [Google Scholar] [PubMed]
- Feng, Y.; Shen, J.; Streaker, E.D.; Lockwood, M.; Zhu, Z.; Low, P.S.; Dimitrov, D.S. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res. Ther. 2011, 13, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and Development of Folic-Acid-Based Receptor Targeting for Imaging and Therapy of Cancer and Inflammatory Diseases. Accounts Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Müller, C. Folate based radiopharmaceuticals for imaging and therapy of cancer and inflammation. Curr. Pharm. Des. 2012, 18, 1058–1083. [Google Scholar] [CrossRef]
- Low, P.S.; Antony, A.C. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv. Drug Deliv. Rev. 2004, 56, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Sega, E.; Low, P. Tumor detection using folate receptor-targeted imaging agents. Cancer Metastasis Rev. 2008, 27, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S.; Kularatne, S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Low, P.S. Folate-targeted therapies for cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef] [PubMed]
- Müller, C. Folate-based radiotracers for PET imaging—Update and perspectives. Molecules 2013, 18, 5005–5031. [Google Scholar] [CrossRef]
- Leamon, C.P.; Reddy, J.A. Folate-targeted chemotherapy. Adv. Drug Deliv. Rev. 2004, 56, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.J.; Lewis, M.R.; Reichert, D.E.; Laforest, R.; Sharp, T.L.; Lewis, J.S.; Yang, Z.; Waters, D.J.; Snyder, P.W.; Low, P.S.; et al. Preparation of 66Ga- and 68Ga-labeled Ga(III)-deferoxamine-folate as potential folate-receptor-targeted PET radiopharmaceuticals. Nucl. Med. Biol. 2003, 30, 725–731. [Google Scholar] [CrossRef]
- Rossin, R.; Pan, D.; Qi, K.; Turner, J.L.; Sun, X.; Wooley, K.L.; Welch, M.J. 64Cu-Labeled folate-conjugated shell cross-linked nanoparticles for tumor imaging and radiotherapy: Synthesis, radiolabeling, and biologic evaluation. J. Nucl. Med. 2005, 46, 1210–1218. [Google Scholar] [PubMed]
- Bettio, A.; Honer, M.; Müller, C.; Brühlmeier, M.; Müller, U.; Schibli, R.; Groehn, V.; Schubiger, A.P.; Ametamey, S.M. Synthesis and preclinical evaluation of a folic acid derivative labeled with 18F for PET imaging of folate receptor-positive tumors. J. Nucl. Med. 2006, 47, 1153–1160. [Google Scholar] [PubMed]
- Ross, T.L.; Honer, M.; Lam, P.Y.H.; Mindt, T.L.; Groehn, V.; Schibli, R.; Schubiger, P.A.; Ametamey, S.M. Fluorine-18 click radiosynthesis and preclinical evaluation of a new 18F-labeled folic acid derivative. Bioconjug. Chem. 2008, 19, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Al Jammaz, I.; Al-Otaibi, B.; Okarvi, S.; Amartey, J. Novel synthesis of [18F]-fluorobenzene and pyridinecarbohydrazide-folates as potential PET radiopharmaceuticals. J. Label. Compd. Radiopharm. 2006, 49, 125–137. [Google Scholar] [CrossRef]
- Al Jammaz, I.; Al-Otaibi, B.; Amer, S.; Okarvi, S.M. Rapid synthesis and in vitro and in vivo evaluation of folic acid derivatives labeled with fluorine-18 for PET imaging of folate receptor-positive tumors. Nucl. Med. Biol. 2011, 38, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Wang, X.; Nicolas, G.; Medina, C.; Raynal, I.; Port, M.; Maecke, H.R. Development of new folate-based PET radiotracers: Preclinical evaluation of 68Ga-DOTA-folate conjugates. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, M.; Tamma, M.-L.; Nicolas, G.P.; Lasri, E.; Medina, C.; Raynal, I.; Port, M.; Weber, W.A.; Maecke, H.R. In vivo imaging of folate receptor positive tumor xenografts using novel 68Ga-NODAGA-folate conjugates. Mol. Pharm. 2012, 9, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Zhernosekov, K.; Köster, U.; Johnston, K.; Dorrer, H.; Hohn, A.; van der Walt, N.T.; Türler, A.; Schibli, R. A unique matched quadruplet of terbium radioisotopes for PET and SPECT and for α- and β--radionuclide therapy: An in vivo proof-of-concept study with a new receptor-targeted folate derivative. J. Nucl. Med. 2012, 53, 1951–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, C.R.; Reber, J.; Müller, A.; Krämer, S.D.; Ametamey, S.M.; Schibli, R.; Müller, C. [18F]Fluoro-deoxy-glucose folate: A novel PET radiotracer with improved in vivo properties for folate receptor targeting. Bioconjug. Chem. 2012, 23, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Al Jammaz, I.; Al-Otaibi, B.; Amer, S.; Al-Hokbany, N.; Okarvi, S. Novel synthesis and preclinical evaluation of folic acid derivatives labeled with 18F-[FDG] for PET imaging of folate receptor-positive tumors. Nucl. Med. Biol. 2012, 39, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gent, Y.Y.J.; Weijers, K.; Molthoff, C.F.M.; Windhorst, A.D.; Huisman, M.C.; Smith, D.E.C.; Kularatne, S.A.; Jansen, G.; Low, P.S.; Lammertsma, A.A.; et al. Evaluation of the novel folate receptor ligand [18F]fluoro-PEG-folate for macrophage targeting in a rat model of arthritis. Arthritis Res. Ther. 2013, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, C.R.; Groehn, V.; Reber, J.; Schibli, R.; Ametamey, S.M.; Müller, C. Improved PET imaging of tumors in mice using a novel 18F-folate conjugate with an albumin-binding entity. Mol. Imaging Biol. 2013, 15, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A.; Bélanger, M.; Meng, X.; Connolly, B.M.; Vanko, A.; Suresch, D.L.; Guenther, I.; Wang, S.; Low, P.S.; Mcquade, P.; et al. Comparative analysis of folate derived PET imaging agents with [18F]-2-fluoro-2-deoxy-D-glucose using a rodent inflammatory paw model. Mol. Pharm. 2013, 10, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Schieferstein, H.; Fischer, C.R.; Ross, T.L.; Betzel, T. 18F-click labeling and preclinical evaluation of a new 18F-folate for PET imaging. EJNMMI Res. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Bunka, M.; Reber, J.; Fischer, C.; Zhernosekov, K.; Türler, A.; Schibli, R. Promises of cyclotron-produced 44Sc as a diagnostic match for trivalent β--emitters: In vitro and in vivo study of a 44Sc-DOTA-folate conjugate. J. Nucl. Med. 2013, 54, 2168–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieferstein, H.; Kelsch, A.; Reibel, A.; Koynov, K.; Barz, M.; Buchholz, H.-G.; Bausbacher, N.; Thews, O.; Zentel, R.; Ross, T.L. 18F-Radiolabeling, preliminary evaluation of folate-pHPMA conjugates via PET. Macromol. Biosci. 2014, 14, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Al Jammaz, I.; Al-otaibi, B.; Al-hokbany, N.; Amer, S.; Okarvi, S. Development and pre-clinical evaluation of new 68Ga-NOTA-folate Conjugates for PET imaging of folate receptor-positive tumors. Anticancer Res. 2014, 34, 6547–6556. [Google Scholar]
- Farkas, R.; Siwowska, K.; Ametamey, S.M.; Schibli, R.; van der Meulen, N.P.; Müller, C. 64Cu- and 68Ga-based PET imaging of folate receptor-positive tumors: Development and evaluation of an albumin-binding NODAGA–folate. Mol. Pharm. 2016, 13, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Mathur, A.; Pandey, U.; Bhatt, J.; Mukherjee, A.; Ram, R.; Sarma, H.D.; Dash, A. Synthesis and evaluation of a 68Ga labeled folic acid derivative for targeting folate receptors. Appl. Radiat. Isot. 2016, 116, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Meng, X.; McQuade, P.; Rubins, D.; Lin, S.; Zeng, Z.; Haley, H.; Miller, P.; Trotter, D.G.; Low, P.S. Synthesis and preclinical evaluation of folate-NOTA-Al18F for PET imaging of folate receptor-positive tumors. Mol. Pharm. 2016, 13, 1520–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boss, S.D.; Betzel, T.; Müller, C.; Fischer, C.R.; Haller, S.; Reber, J.; Groehn, V.; Schibli, R.; Ametamey, S.M. Comparative studies of three pairs of α- and γ-conjugated folic acid derivatives labeled with fluorine-18. Bioconjug. Chem. 2016, 27, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.; Longo, V.A.; Groaning, M.; Weber, W.A.; Reiner, T. Development of a new folate-derived Ga-68-based PET imaging agent. Mol. Imaging Biol. 2017, 19, 754–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Meng, X.; Mcquade, P.; Rubins, D.; Lin, S.; Zeng, Z.; Haley, H.; Miller, P.; Gonza, D.G.; Low, P.S. Folate-PEG-NOTA-Al18F: A new folate based radiotracer for PET imaging of folate receptor-positive tumors. Mol. Pharm. 2017, 14, 4353–4361. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Fu, F.; Zhu, J.; Huang, R.; Zhu, Y.; Liu, Z.; Wang, J.; Conti, P.S.; Shi, X.; Chen, K. 64Cu-Labeled multifunctional dendrimers for targeted tumor PET imaging. Nanoscale 2018, 10, 6113–6124. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, K.; Reffert, L.M.; Schieferstein, H.; Pektor, S.; Eckert, R.; Miederer, M.; Rösch, F.; Ross, T.L. Comparison study of two differently clicked 18F-folates—Lipophilicity plays a key role. Pharmaceuticals 2018, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, P.S.; Lee, J.Y.; Park, J.H.; Kim, S.W. Synthesis and evaluation of 68Ga-HBED-CC-EDBE-folate for positron-emission tomography imaging of overexpressed folate receptors on CT26 tumor cells. J. Label. Compd. Radiopharm. 2018, 61, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Radford, L.L.; Fernandez, S.; Beacham, R.; El Sayed, R.; Farkas, R.; Benešov, M.; Müller, C.; Lapi, S.E. New 55Co-labeled albumin-binding folate derivatives as potential PET agents for folate receptor imaging. Pharmaceuticals 2019, 12, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, G.S.; Detering, L.; Luehmann, H.P.; Primeau, T.; Lee, Y.-S.; Laforest, R.; Li, S.; Stec, J.; Lim, K.-H.; Lockhart, A.C.; et al. Folate receptor α-targeted 89Zr-M9346A immuno-PET for image-guided intervention with mirvetuximab soravtansine in triple-negative breast cancer. Mol. Pharm. 2019, 16, 3996–4006. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.-B.; Moon, M.M.; Chellan, J.R.; Hwang, S.H.; Lee, J.H.; Jung, S.J.; Kim, B.C.; Yu, K.H. In vitro PET/MRI diagnosis and targeted chemotherapy for cancer using radiolabeled nanoprobe: A theragnostic approach. Korean Chem. Soc. 2016, 37, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Cho, B.-B.; Anusha, J.R.; Jung, S.; Raj, C.J.; Kim, B.C.; Yu, K.H. Synthesis of 64Cu-radiolabeled folate-conjugated iron oxide nanoparticles for cancer diagnosis. J. Nanosci. Nanotechnol. 2020, 20, 2040–2044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, Q.; He, Y.; Zhang, C.; Zhu, H.; Yang, Z.; Lu, J. Synthesis and biological evaluation of 68Ga-labeled pteroyl-Lys conjugates for folate receptor-targeted tumor imaging. J. Label. Compd. Radiopharm. 2016, 59, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Hohn, A.; Schubiger, P.A.; Schibli, R. Preclinical evaluation of novel organometallic 99mTc-folate and 99mTc-pteroate radiotracers for folate receptor-positive tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guo, H.; Xie, F.; Lu, J. Preparation and biological evaluation of 99mTcN-labeled pteroyl-lys derivative as a potential folate receptor imaging agent. J. Label. Compd. Radiopharm. 2014, 57, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.Y.; Mathias, C.J.; Green, M.A. Targeting the tumor-associated folate receptor with an 111In-DTPA conjugate of pteroic acid. J. Am. Chem. Soc. 2005, 127, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xie, F.; Zhu, M.; Li, Y.; Yang, Z.; Wang, X.; Lu, J. The synthesis of pteroyl-lys conjugates and its application as technetium-99m labeled radiotracer for folate receptor-positive tumor targeting. Bioorg. Med. Chem. Lett. 2011, 21, 2025–2029. [Google Scholar] [CrossRef]
- Ross, T.L.; Müller, C.; Groehn, V.; Schibli, R.; Ametamey, S.M. A new 18F-labeled folic acid derivative with improved properties for the PET imaging of folate receptor-positive tumors. J. Nucl. Med. 2010, 51, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Betzel, T.; Müller, C.; Groehn, V.; Müller, A.; Reber, J.; Fischer, C.R.; Krämer, S.D.; Schibli, R.; Ametamey, S.M. Radiosynthesis and preclinical evaluation of 3′-aza-2′-[18F]fluorofolic acid: A novel PET radiotracer for folate receptor targeting. Bioconjug. Chem. 2013, 24, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Tewson, T.J.; Erdahl, C.E.; Kohen, A. A fast chemoenzymatic synthesis of [11C]-N5,N10-methylenetetrahydrofolate as a potential PET tracer for proliferating cells. Nucl. Med. Biol. 2012, 39, 697–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaitilingam, B.; Chelvam, V.; Kularatne, S.A.; Poh, S.; Ayala-Lopez, W.; Low, P.S. A folate receptor-α-specific ligand that targets cancer tissue and not sites of inflammation. J. Nucl. Med. 2012, 53, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boss, S.D.; Müller, C.; Siwowska, K.; Büchel, J.I.; Schmid, R.M.; Groehn, V.; Schibli, R.; Ametamey, S.M. Reduced 18F-folate conjugates as a new class of PET tracers for folate receptor imaging. Bioconjug. Chem. 2018, 29, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Boss, S.D.; Müller, C.; Siwowska, K.; Schmid, R.M.; Groehn, V.; Schibli, R.; Ametamey, S.M. Diastereomerically pure 6R- and 6S-3′-Aza-2′-18F-fluoro-5-methyltetrahydrofolates show unprecedentedly high uptake in folate receptor-positive KB tumors. J. Nucl. Med. 2019, 60, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Yong, E.-L.; Xu, H.E.; Melcher, K. Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500, 486–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackman, A.L.; Theti, D.S.; Gibbs, D.D. Antifolates targeted specifically to the folate receptor. Adv. Drug Deliv. Rev. 2004, 56, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.-Y.; Mathias, C.J.; Green, M.A. Folate-receptor-targeted radionuclide imaging agents. Adv. Drug Deliv. Rev. 2004, 56, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.V.; Antich, P.P.; Constantinescu, A.; Anderson, J.A.; Fernando, J.L.; Prior, J.; Nguyen, T.; Parkey, R.W.; Weitman, S.D.; Kamen, B.A. Imaging of folate receptor with I-125 labeled folate using small animal imaging system built with plastic scintillating optical fibers. SPIE 1994, 2281, 76–81. [Google Scholar]
- Ke, C.-Y.; Mathias, C.J.; Green, M.A. The folate receptor as a molecular target for tumor-selective radionuclide delivery. Nucl. Med. Biol. 2003, 30, 811–817. [Google Scholar] [CrossRef]
- Taylor, J. The diagnostic application of radiolabelled folate in the detection of folate receptor-positive tumors. J. Med. Imaging Radiat. Sci. 2014, 45, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Schibli, R. Folic acid conjugates for nuclear imaging of folate receptor-positive cancer. J. Nucl. Med. 2011, 52, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Lee, R.J.; Mathias, C.J.; Green, M.A.; Low, P.S. Synthesis, purification, and tumor cell uptake of 67Ga-deferoxamine-folate, a potential radiopharmaceutical for tumor imaging. Bioconjug. Chem. 1996, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Siwowska, K.; Schmid, R.; Cohrs, S.; Schibli, R.; Müller, C. Folate receptor-positive gynecological cancer cells: In vitro and in vivo characterization. Pharmaceuticals 2017, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Struthers, H.; Winiger, C.; Zhernosekov, K.; Schibli, R. DOTA Conjugate with an albumin-binding entity enables the first folic acid-targeted 177Lu-radionuclide tumor therapy in mice. J. Nucl. Med. 2013, 54, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verweij, N.J.F.; Yaqub, M.; Bruijnen, S.T.G.; Pieplenbosch, S.; TerWee, M.M.; Jansen, G.; Chen, Q.; Low, P.S.; Windhorst, A.D.; Lammertsma, A.A.; et al. First in man study of [18F]fluoro-PEG-folate PET: A novel macrophage imaging technique to visualize rheumatoid arthritis. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Choi, N.; Hwang, S.; Yim, M.S.; Lee, J.-S.; Lee, S.-M.; Cho, G.; Ryu, E.K. Folate receptor-specific positron emission tomography imaging with folic acid-conjugated tissue inhibitor of metalloproteinase-2. Bull. Korean Chem. Soc. 2013, 34, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Ilgan, S.; Yang, D.J.; Higuchi, T.; Zareneyrizi, F.; Bayhan, H.; Yu, D.; Kim, E.E.; Podoloff, D.A. 99mTc-Ethylenedicysteine-Folate: A new tumor imaging agent. Synthesis, labeling and evaluation in animals. Cancer Biother. Radiopharm. 1998, 13, 427–436. [Google Scholar] [PubMed]
- Leamon, C.P.; Parker, M.A.; Vlahov, I.R.; Xu, L.-C.; Reddy, J.A.; Vetzel, M.; Douglas, N. Synthesis and biological evaluation of EC20: A new folate-derived, 99mTc-based radiopharmaceutical. Bioconjug. Chem. 2002, 13, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.E.; Siegel, B.A.; Edell, S.L.; Oyesiku, N.M.; Morgenstern, D.E.; Messmann, R.A.; Amato, R.J. Exploratory study of 99mTc-EC20 imaging for identifying patients with folate receptor-positive solid tumors. J. Nucl. Med. 2008, 49, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Nakatani, H.; Yanaihara, H.; Omote, M. Phase I clinical trial of 99mTc-etarfolatide, an imaging agent for folate receptor in healthy Japanese adults. Ann. Nucl. Med. 2015, 29, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.T.; Joyrich, R.N.; Naumann, R.W.; Shah, N.P.; Maurer, A.H.; Strauss, H.W.; Uszler, J.M.; Symanowski, J.T.; Ellis, P.R.; Harb, W.A. Phase II study of treatment of advanced ovarian cancer with folate-receptor-targeted therapeutic (vintafolide) and companion SPECT-based imaging agent (99mTc-etarfolatide). Ann. Oncol. 2014, 25, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Van Der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, N.; Solbach, C.; Voelter, W.; Machulla, H.-J. Nucleophilic aromatic substitution by [18F]fluoride at substituted 2-nitropyridines. J. Radioanal. Nucl. Chem. 2009, 283, 757–764. [Google Scholar] [CrossRef]
- Haberkorn, U.; Mier, W.; Kopka, K.; Herold-Mende, C.; Altmann, A.; Babich, J. Identification of ligands and translation to clinical applications. J. Nucl. Med. 2017, 58, 27S–33S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisel, A.; Burger, I.; Müller, J.; Gnesin, S.; Siano, M.; Früh, M.; Müller, C.; Geistlich, S.; Heafliger, L.; Ametamey, S.M.; et al. First-in-Human Study with 18F-Azafol for Folate Receptor Targeted PET: An Interim-Analysis of the PET_FOL_1 Phase-I Trial. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, S219. [Google Scholar]
- Gnesin, S.; Müller, J.; Burger, I.A.; Meisel, A.; Siano, M.; Früh, M.; Choschzick, M.; Müller, C.; Schibli, R.; Ametamey, S.M.; et al. Radiation dosimetry of 18F-AzaFol: A first in-human use of a folate receptor PET tracer. EJNMMI Res. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.; Douglas, K.; Roller, L.; Fisher, A.; Peterson, T.; Liu, F.; Nickels, M.; Smith, G.; Blackwell, T.; Manning, H.C. First-in-human PET imaging study using [68Ga]-folate tracer, [68Ga]EC2115. J. Nucl. Med. 2019, 60, 1062. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Year | Radiolabeled Folate | Main Findings | Ref. |
|---|---|---|---|---|
| Pendant Approach (FA-Based Derivatives) | ||||
| Mathis et al. | 2003 | α/γ-[66Ga]/[68Ga]Ga-deferoxamine-folate | High kidney and abdominal uptake | [22] |
| Rossin et al. | 2005 | 64Cu[Cu]TETA-SCK-folate | Tumor uptake of 5.9% IA/g and high liver uptake of 33.8% IA/g at 4 h p.i. (post injection) | [23] |
| Bettio et al. | 2006 | α/γ-[18F]Fluorobenzylamine-folate (1) | High abdominal uptake; Tu (tumor): 6.56% IA/g, Ki (kidney): 40.7% IA/g at 125 min p.i. | [24] |
| Ross et al. | 2008 | γ-Click-[18F]fluorobutyl-folate (2) | High abdominal uptake; Tu: 3.13% IA/g, Ki: 16.5% IA/g at 45 min p.i. | [25] |
| Al Jammaz et al. | 2006, 2011 | [18F]fluorobenzene- and [18F]fluoropyridine-carbohydrazide-folate (6)/methotrexate | Tumor uptake < 6% IA/g, rapid blood clearance | [26,27] |
| Fani et al. | 2011 | [68Ga]Ga-DOTA folate conjugates | Tumor uptake of around 12% IA/g, high kidney uptake of 56% IA/g 60 min p.i. | [28] |
| Fani et al. | 2012 | [68Ga]Ga-NODAGA-folic acid (9) and -5,8-dideazafolic acid conjugates | FA derivative outperformed the 5,8-dideazafolic acid derivative, fast blood clearance, high kidney uptake (Tu: 16.6% IA/g, Ki: 91.5% IA/g 60 min p.i.) | [29] |
| Müller et al. | 2012 | [152Tb]Tb-cm09 with DOTA chelator (10) | Clear visualization of tumor xenografts and kidneys at 24 h p.i. | [30] |
| Fischer et al. | 2012 | γ-Click-[18F]fluoro-deoxy-glucose-folate (3) | High liver uptake; Tu: 10.0% IA/g, Ki: 42.9% IA/g at 60 min p.i. | [31] |
| Al Jammaz et al. | 2012 | [18F]FDG-folate/methotrexate | Tumor uptake < 4% IA/g, rapid blood clearance | [32] |
| Gent et al. | 2013 | [18F]Fluoro-PEG-folate (7) | Targeting of activated macrophages in rat model of arthritis | [33] |
| Fischer et al. | 2013 | Album-binding [18F]FDG-folate (4) | Tu: 11.5% IA/g, Ki: 13.4% IA/g at 60 min p.i.; Tu: 15.2% IA/g, Ki: 18.1% IA/g at 4 h p.i. | [34] |
| Kularatne et al. | 2013 | 4-[18F]Fluorophenyl- and [68Ga]Ga-DOTA-folates | Targeting of activated macrophages in rodent inflammatory paw model | [35] |
| Schieferstein et al. | 2013 | [18F]Oligoethyleneglycol-folate | Tu: 3.39% IA/g; Ki: 40.8% IA/g at 60 min p.i. | [36] |
| Müller et al. | 2013 | [44Sc]Sc-cm09 with DOTA chelator (11) | Favorable tissue distribution with low uptake in non-targeted tissues Tu: 8.37% IA/g, Ki: 19.2% IA/g at 2h p.i.; Tu: 14.1% IA/g, Ki: 22.2% IA/g at 20 h p.i. | [37] |
| Schieferstein et al. | 2014 | [18F]-Labeled folate-pHPMA | High kidney uptake and low tumor uptake of < 0.5% IA/g at 2 and 4 h p.i. | [38] |
| Al Jammaz et al. | 2014 | [68Ga]Ga-NOTA- and -Ga-NOTAM-folate conjugates | Moderate uptake in the kidneys and liver; NOTA-folate: Tu: 17.8% IA/g 4 h p.i.; NOTAM-folate: Tu: 8.65% IA/g 4 h p.i. | [39] |
| Farkas et al. | 2016 | [64Cu]Cu- and [68Ga]Ga-rf42 with albumin-binding entity and NODAGA chelator (12 and 13) | More promising features of [64Cu]-rf42, high tumor-to-kidney ratio of 0.73. Tu: 13.4% IA/g, Ki: 25.3% IA/g at 2 h p.i.; Tu: 16.2% IA/g, Ki: 29.5% IA/g at 20 h p.i. | [40] |
| Jain et al. | 2016 | [68Ga]Ga-NOTA-folic acid | No in vivo results with FR-positive tumor-bearing mice reported | [41] |
| Chen et al. | 2016 | [18F]Folate-NOTA-AlF (8) | Low liver uptake; Tu: 10.9% IA/g, Ki: 78.6% IA/g at 90 min p.i. | [42] |
| Boss et al. | 2016 | α- and γ-Click-[18F]fluorobutyl-folates (2), -[18F]fluoroethyl-folates (5), and -[18F]FDG-folates (3) | α-isomers show significant increased kidney uptake, γ-isomers significantly higher liver uptake (see publication for biodistribution results) | [43] |
| Brand et al. | 2017 | [68Ga]Ga-NOTA-PEG-folate | Moderate tumor and kidney uptake, low liver uptake (<1% IA/g at 4.5 h p.i.) Tu: 6.61% IA/g, Ki: 21.7% IA/g at 4.5 h p.i. | [44] |
| Chen et al. | 2017 | Folate-PEG-NOTA-Al[1 8F] | Low liver uptake; Tu: 9.20% IA/g, Ki: 55.3% IA/g at 90 min p.i. | [45] |
| Ma et al. | 2018 | [64Cu]Cu-DOTA-FA-FI-G5·NHAc dendrimers | High liver uptake; Tu: 7.02% IA/g, Ki: 9.81% IA/g at 4 h p.i. | [46] |
| Kettenbach et al. | 2018 | [18F]DBCO- and [18F]Ala-folates | High kidney and abdominal uptake; tumor uptake < 2% IA/g at 60 min p.i. | [47] |
| Choi et al. | 2018 | [68Ga]Ga-HBED-CC-EDBE-folate | No biodistribution data, unfavorable PET imaging due to high abdominal uptake | [48] |
| Radford et al. | 2019 | [55Co]Co-cm10 and [55Co]Co-rf42 (14) | No improvement in tumor-to-kidney ratio compared to 64Cu-labeled derivatives, but lower liver uptake; tumor uptake is 17% IA/g at 4 h for both tracers. | [49] |
| Heo et al. | 2019 | 89Zr[Zr]M9346A | Imaging agent for therapeutic agent IMGN853, which is currently in clinical trials; promising biological results, however, further studies needed | [50] |
| Cho et al. Park et al. | 2016 2020 | [68Ga]Ga(NF)HFCNP [64Cu]Cu-FANCFe | Specific uptake in FR-positive cells, no in vivo data available | [51] [52] |
| Zhang et al. Müller et al. Chen et al. Ke et al. Guo et al. | 2005–2016 | Pteroyl-conjugates | Low tumor and high kidney uptake | [53,54,55,56,57] |
| Integrated Approach (FA-Based Derivatives) | ||||
| Ross et al. | 2010 | 2′-[18F]fluorofolic acid ([18F]FFA, 15) | High abdominal and liver uptake; Tu: 9.37% IA/g, Ki: 46.1% IA/g at 75 min p.i. | [58] |
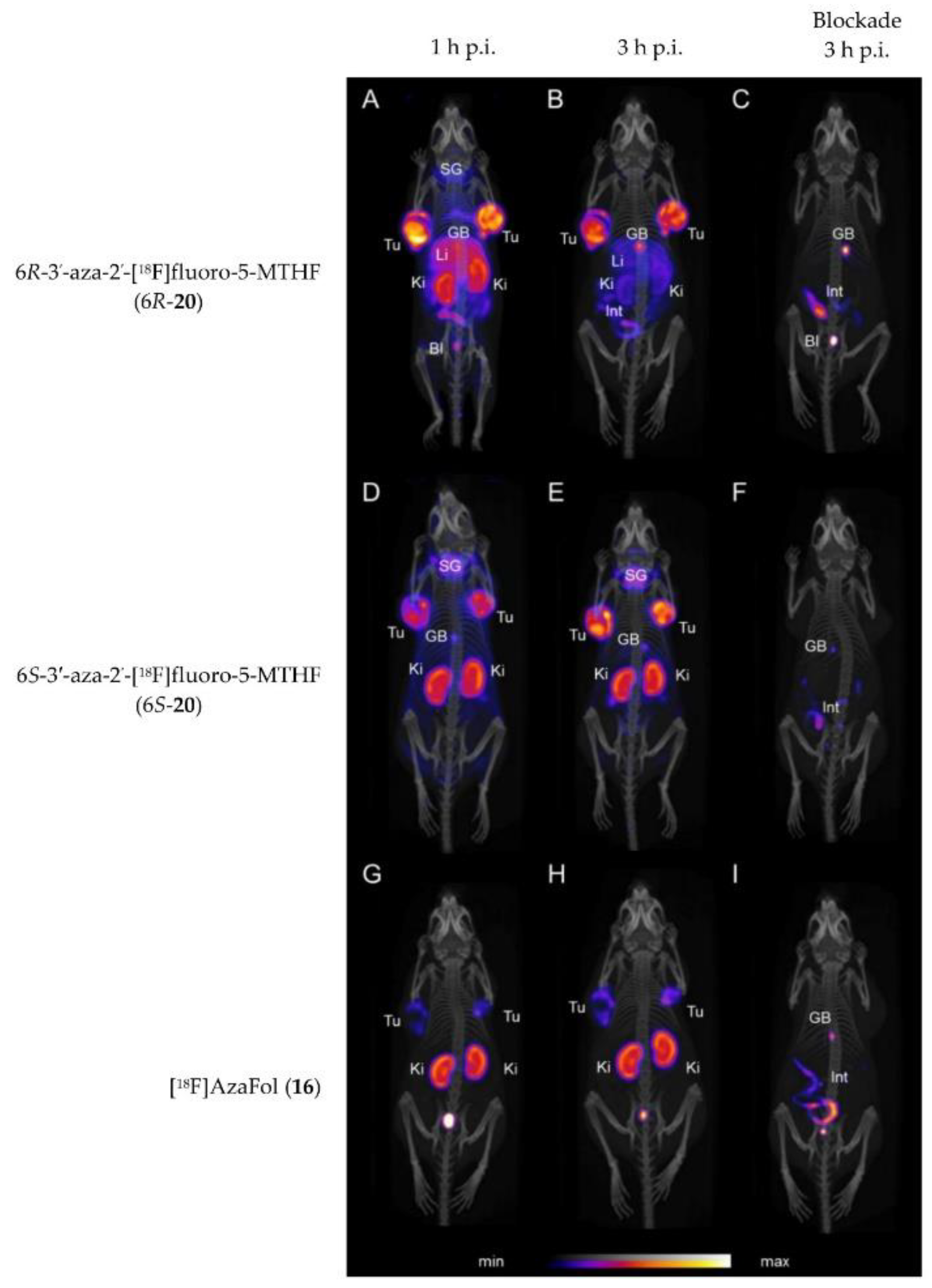
| Betzel et al. | 2013 | 3′-aza-2′-[18F]fluorofolic acid ([18F]AzaFol, 16) | High liver uptake; Tu: 12.6% IA/g, Ki: 57.3% IA/g at 90 min p.i. | [59] |
| Reduced Folate Derivatives | ||||
| Saeed et al. | 2012 | [11C]-N5,N10-methylenetetrahydrofolate (17) | No biological results reported | [60] |
| Vaitilingam et al. | 2012 | [99mTc]Tc-DMTHF (18) | Prove of selectivity of the radiotracer to FR-α over FR-β | [61] |
| Boss et al. | 2018 | 6S-α, 6S-γ, 6R-α, and 6R-γ-click-[18F]fluoroethyl-5-MTHF (6S-α-, 6S-γ-, 6R-α-, and 6R-γ-19) | Different uptake between R- and S-isomers in various organs including kidneys and liver. Tumor uptake between 8–11% IA/g at 60 min p.i. | [62] |
| Boss et al. | 2019 | 6R- and 6S-3′-aza-2′-[18F]fluoro-5-MTHF (6R-20 and 6S-20) | Tumor uptake of over 32% IA/g for both isomers at 3 h p.i. Different uptake between R- and S-isomers in various organs including kidney and liver. | [63] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boss, S.D.; Ametamey, S.M. Development of Folate Receptor−Targeted PET Radiopharmaceuticals for Tumor Imaging—A Bench-to-Bedside Journey. Cancers 2020, 12, 1508. https://doi.org/10.3390/cancers12061508
Boss SD, Ametamey SM. Development of Folate Receptor−Targeted PET Radiopharmaceuticals for Tumor Imaging—A Bench-to-Bedside Journey. Cancers. 2020; 12(6):1508. https://doi.org/10.3390/cancers12061508
Chicago/Turabian StyleBoss, Silvan D., and Simon Mensah Ametamey. 2020. "Development of Folate Receptor−Targeted PET Radiopharmaceuticals for Tumor Imaging—A Bench-to-Bedside Journey" Cancers 12, no. 6: 1508. https://doi.org/10.3390/cancers12061508
APA StyleBoss, S. D., & Ametamey, S. M. (2020). Development of Folate Receptor−Targeted PET Radiopharmaceuticals for Tumor Imaging—A Bench-to-Bedside Journey. Cancers, 12(6), 1508. https://doi.org/10.3390/cancers12061508