Human Plasma-Derived 3D Cultures Model Breast Cancer Treatment Responses and Predict Clinically Effective Drug Treatment Concentrations

 ,
, 
Abstract
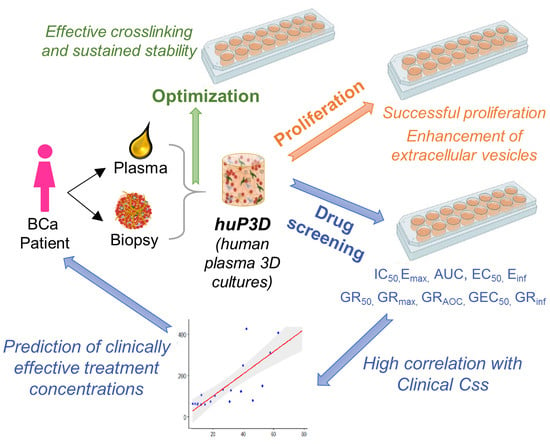
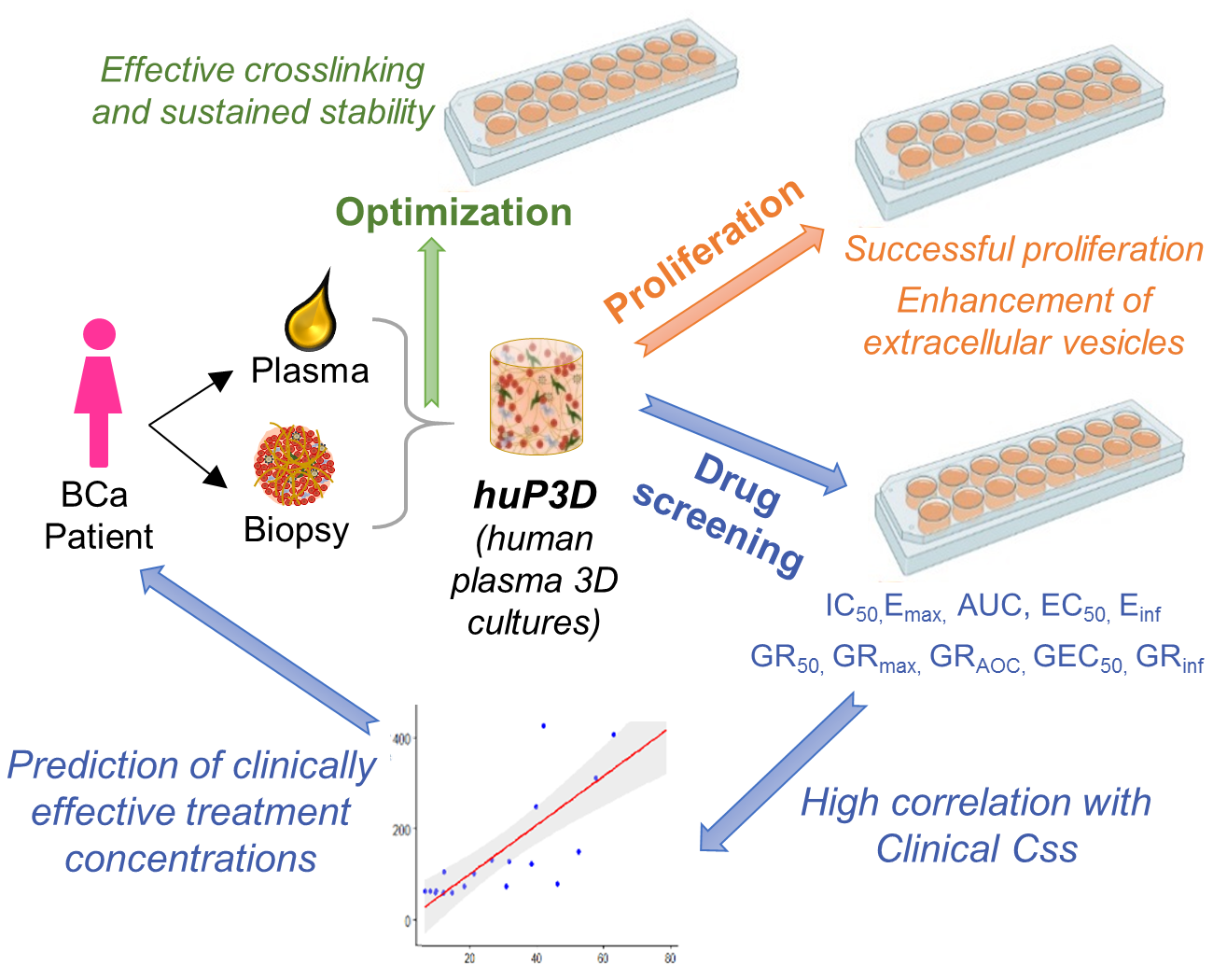
:
1. Introduction
2. Results
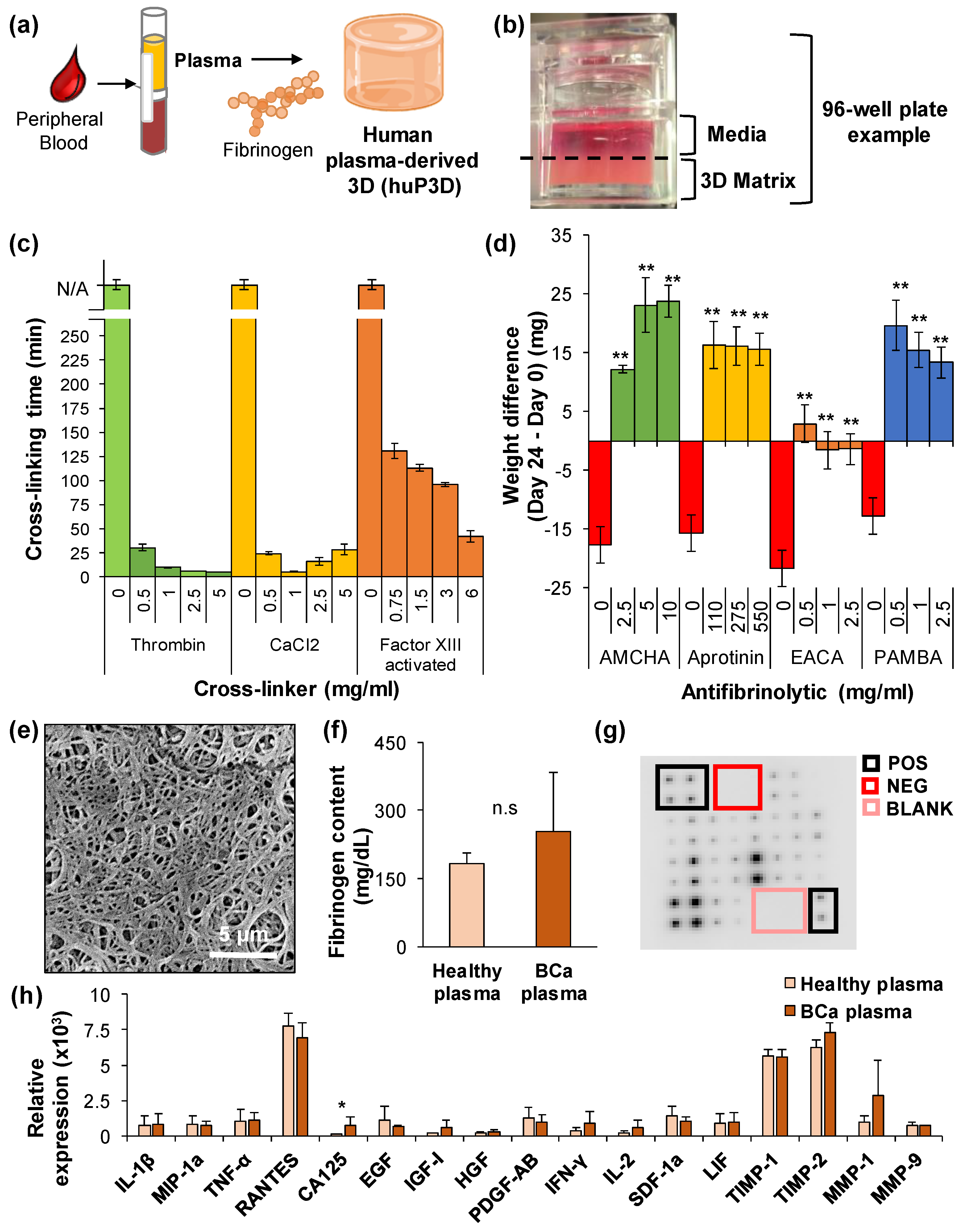
2.1. Chemical and Physical Characterization of the HuP3D Culture Model
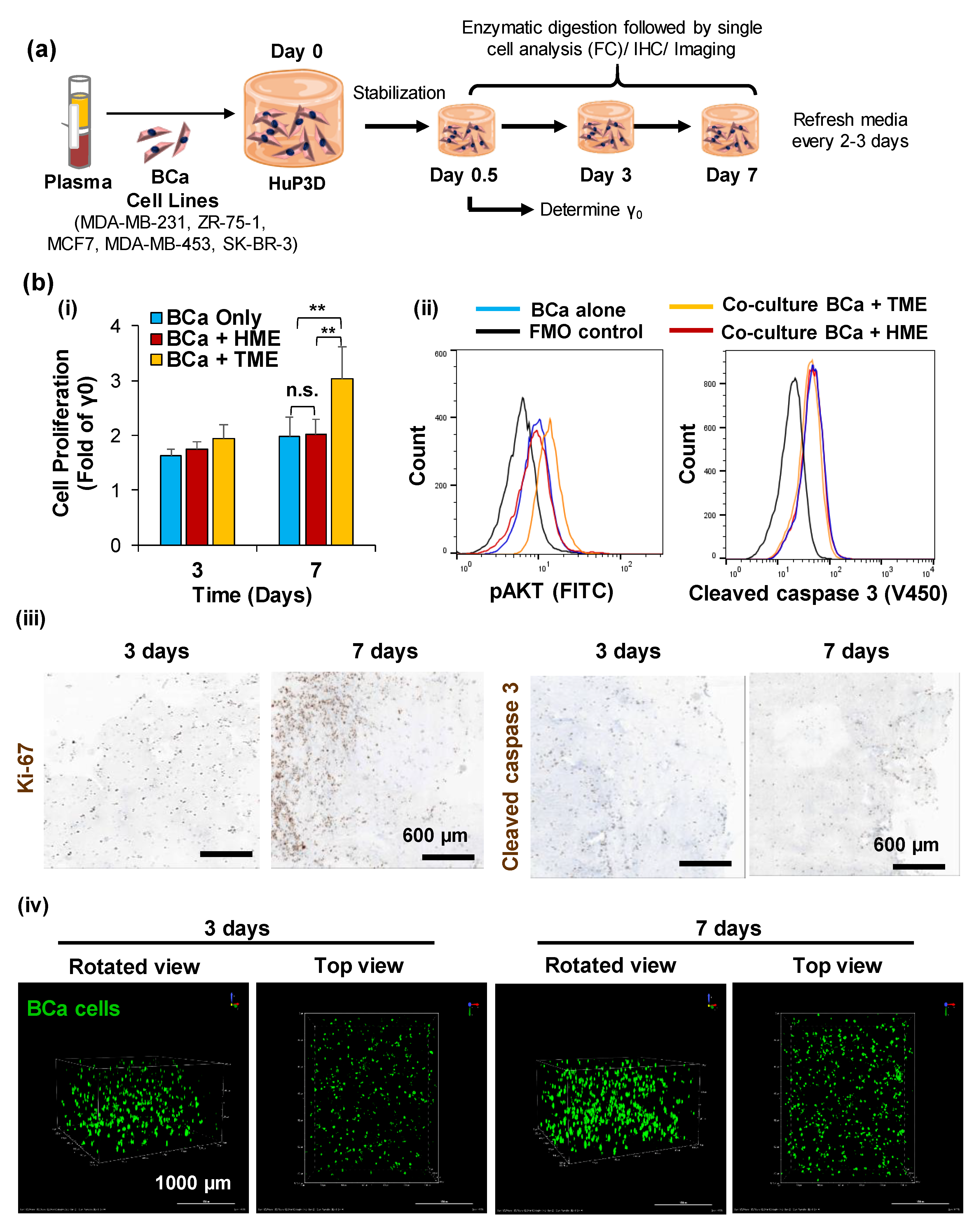
2.2. HuP3D Culture Supports BCa Proliferation
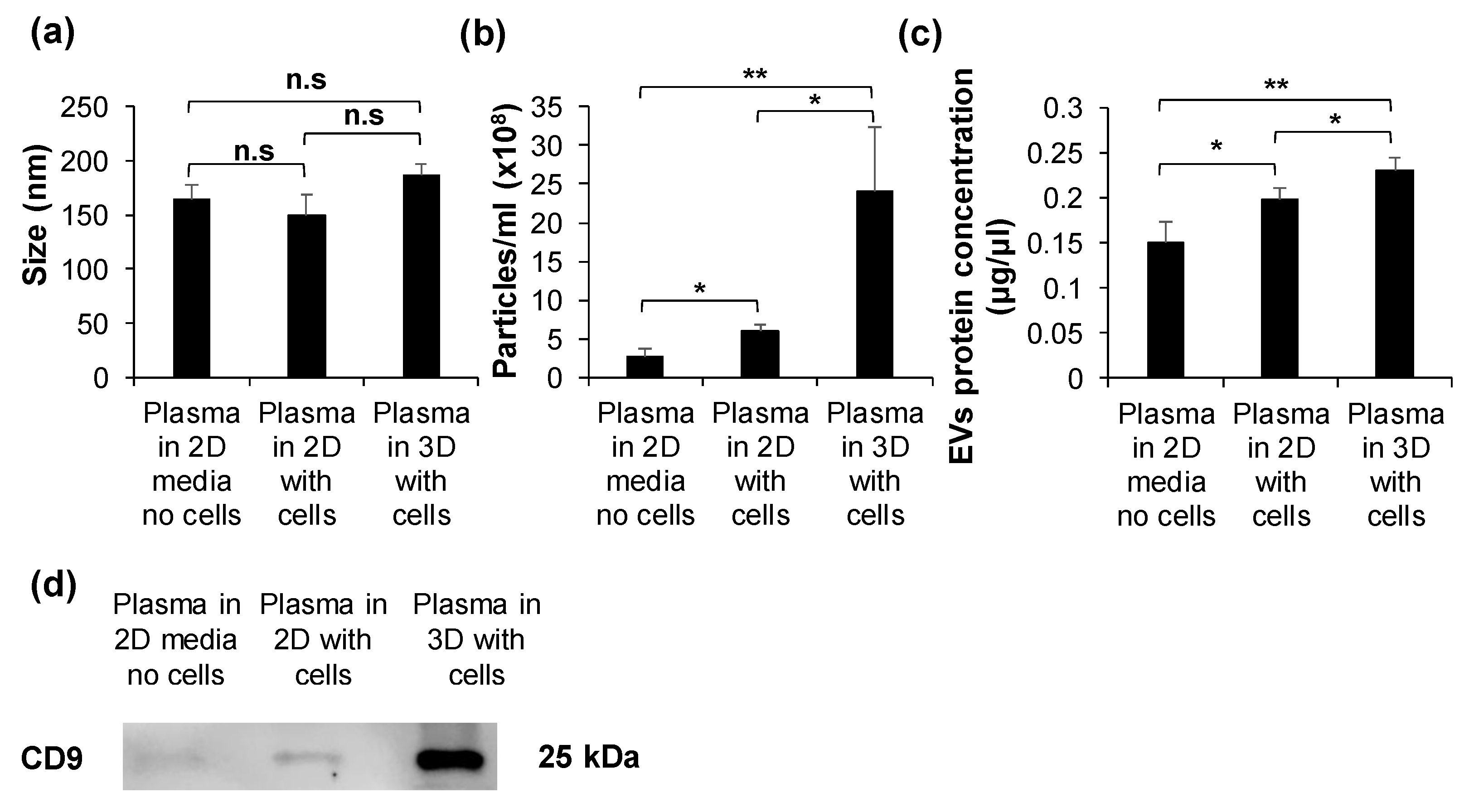
2.3. HuP3D Culture Enhances Extracellular Vesicle (EV) Secretion
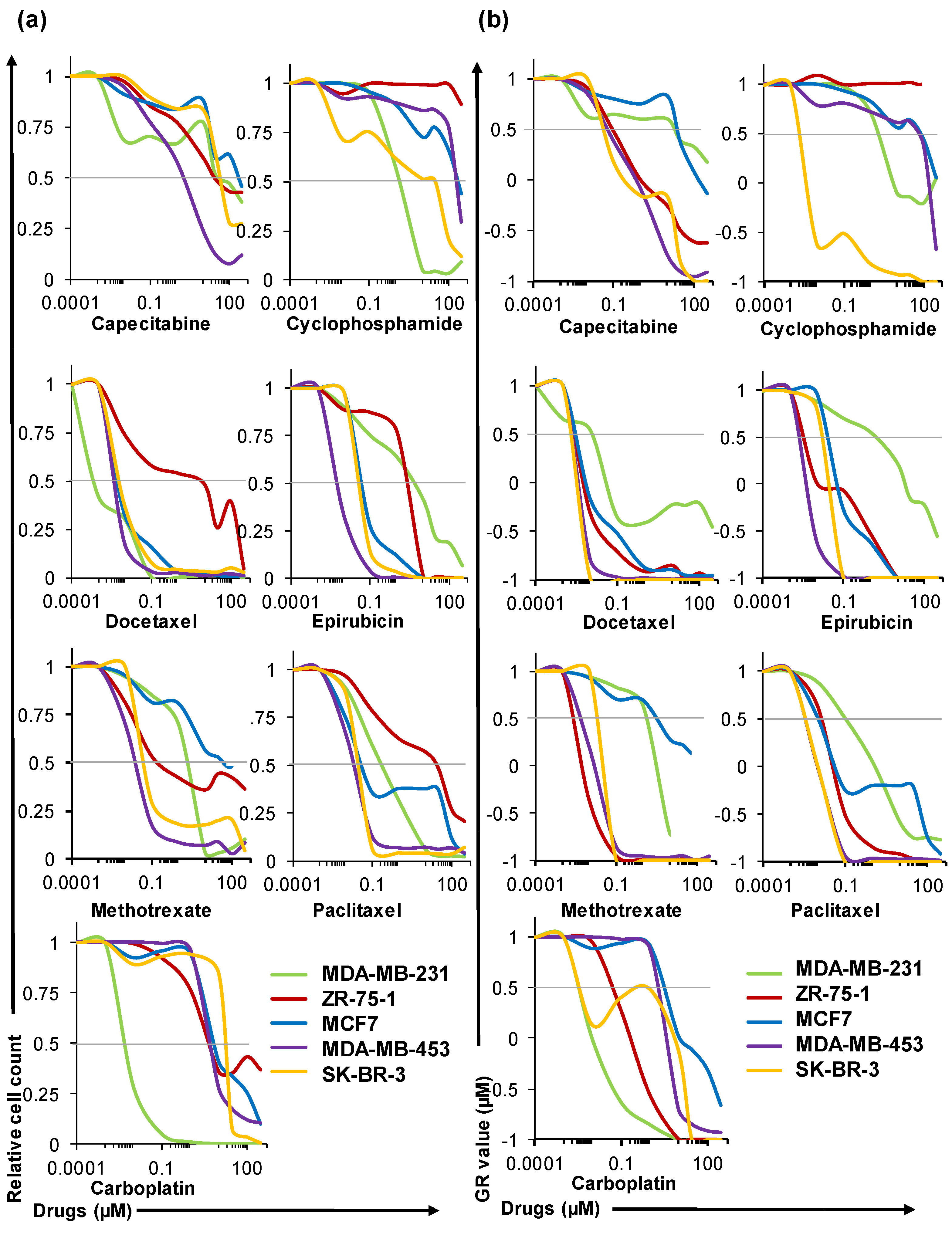
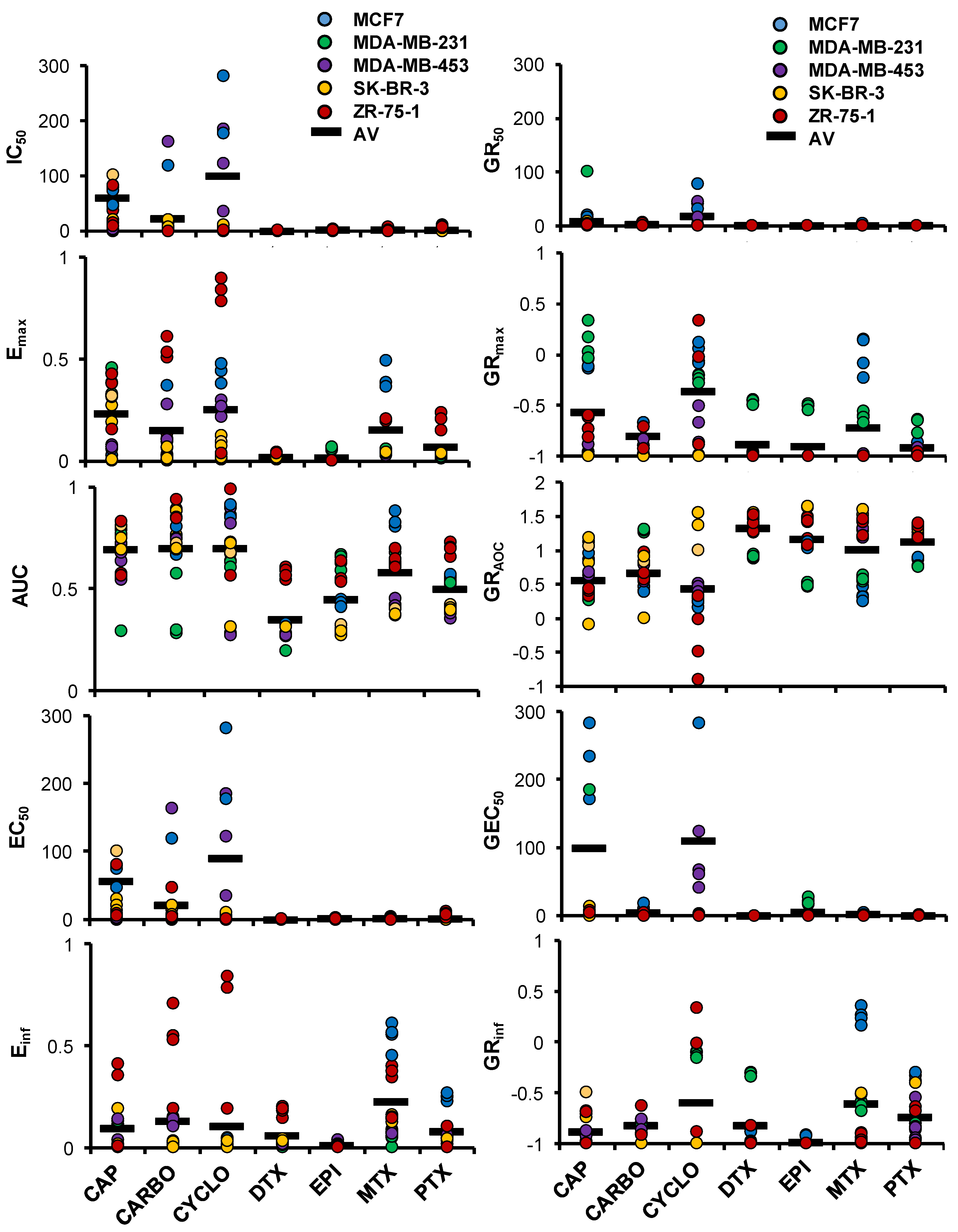
2.4. HuP3D Cultures Allow High-Throughput Drug Screening of BCa Cell Lines
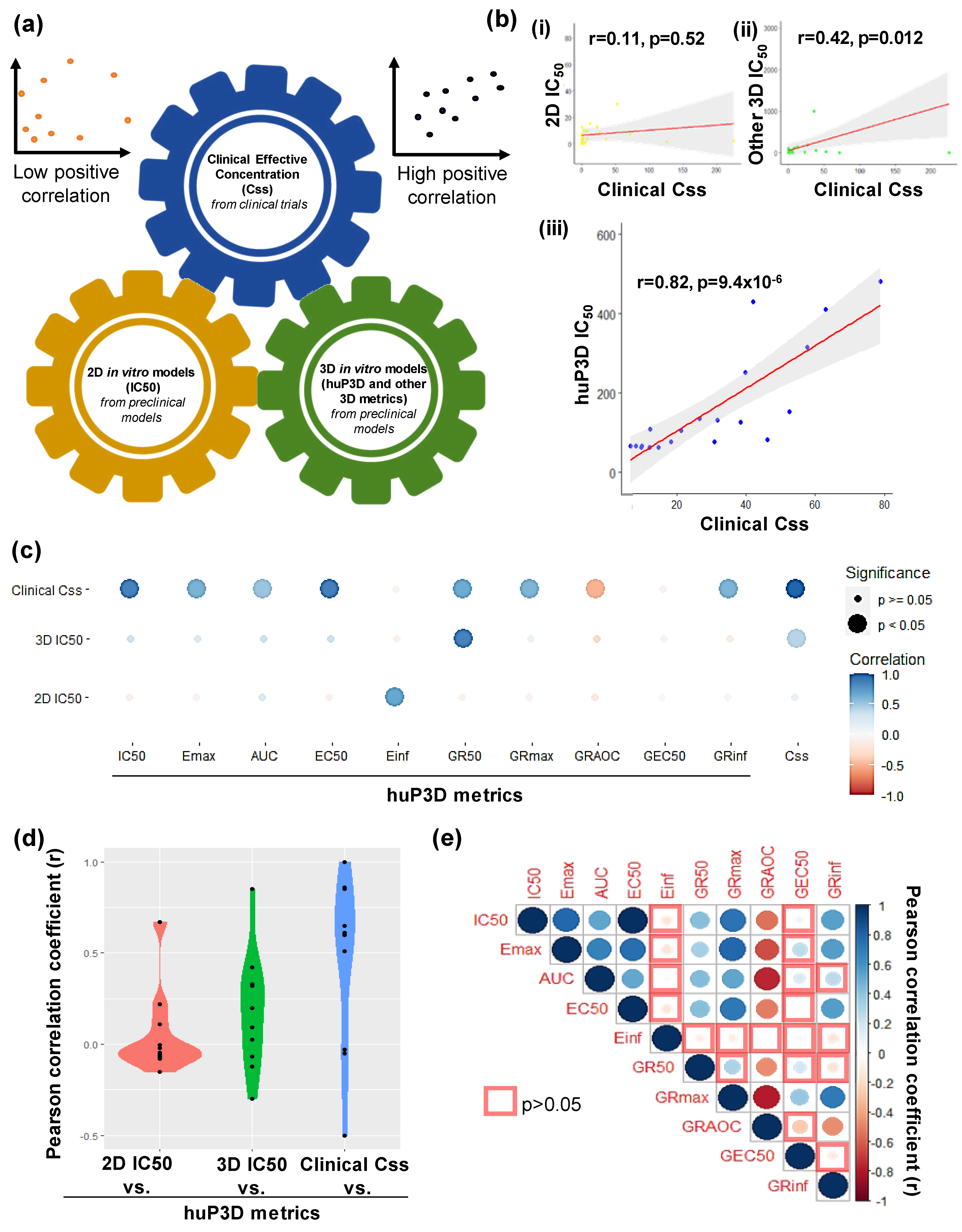
2.5. HuP3D Culture Drug Metrics Correlate Better Than Other In Vitro Models with Clinical Data
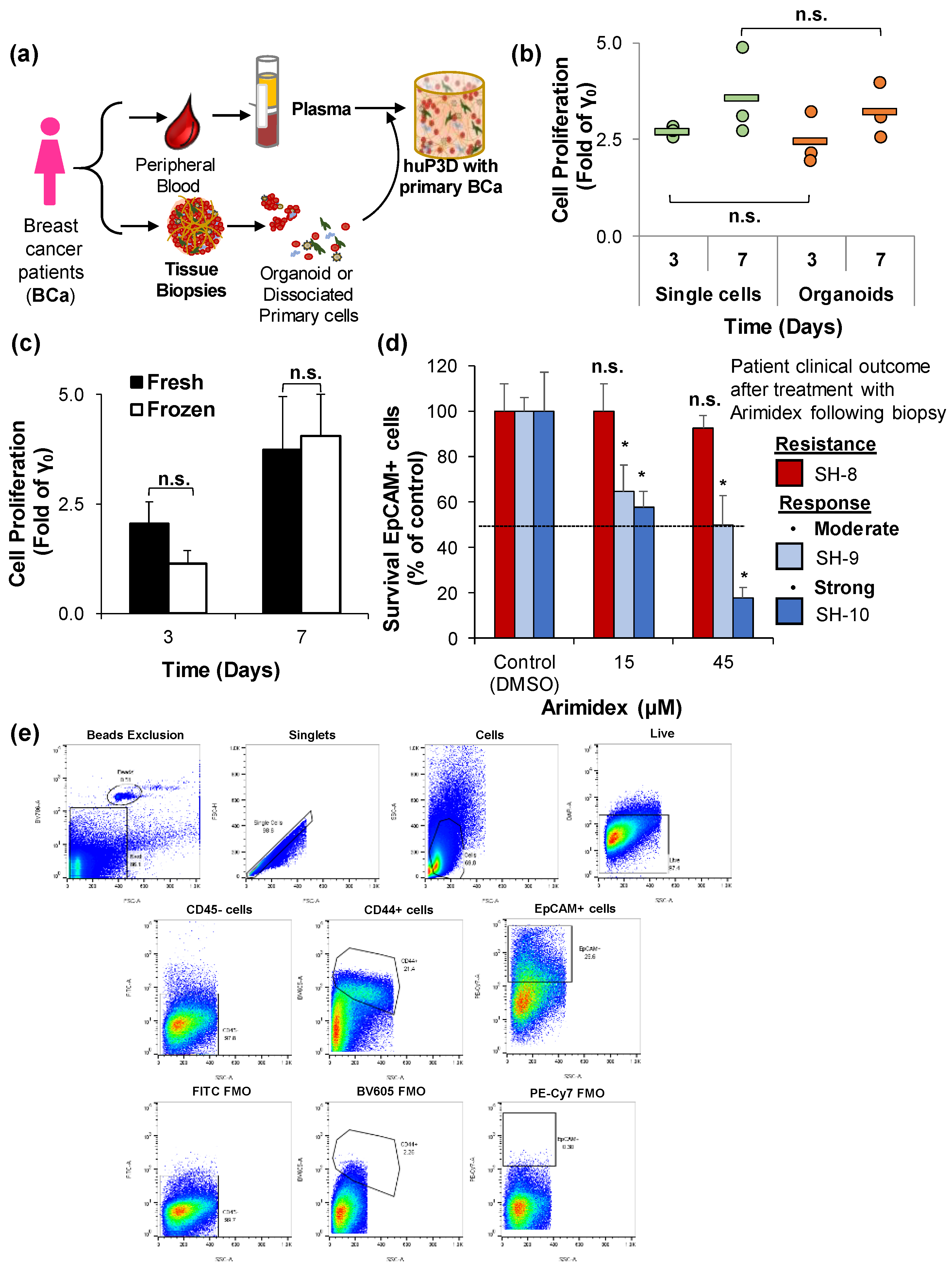
2.6. HuP3D Cultures Support Primary BCa Proliferation and Retrospective Prediction of Therapeutic Efficacy in BCa Patients
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Human Samples
4.4. Development and Characterization of HuP3D Cultures
4.5. Fibrinogen Content in Plasma
4.6. Cytokine Expression in HuP3D Cultures
4.7. Proliferation Analysis by Flow Cytometry of BCa Cell Lines
4.8. Cell Proliferation and Apoptosis Signaling
4.9. Immunohistochemistry Studies (IHC)
4.10. Confocal Imaging and Analysis
4.11. Extracellular Vesical Isolation and Corresponding Vesical Characterization
4.12. Dynamic Light Scattering (DLS) Analysis
4.13. Western Blot
4.14. Drug Screening of BCa Cell Lines in HuP3D Cultures Analyzed by Flow Cytometry
4.15. Determining Drug Metrics Using the GR Calculator
4.16. Correlation of Drug Metrics
4.17. Proliferation and Drug Screening Analysis by Flow Cytometry of Primary BCa Cells
4.18. Statistical Analysis of Data
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibrahim, E.; Al-Gahmi, A.M.; Zeenelin, A.A.; Zekri, J.M.; Elkhodary, T.R.; Gaballa, H.E.; Fawzy, E.E.; El sayed, M.E.; Alzahrani, M.S. Basal vs. luminal A breast cancer subtypes: A matched case-control study using estrogen receptor, progesterone receptor, and HER-2 as surrogate markers. Med. Oncol. 2009, 26, 372–378. [Google Scholar] [CrossRef]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2010, 220, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Kreike, B.; Reis-Filho, J.S. Metaplastic breast carcinomas are basal-like breast cancers: A genomic profiling analysis. Breast Cancer Res. Treat. 2009, 117, 273–280. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Bray, F.; Ferlay, J.; Lortet-Tieulent, J.; Anderson, B.O.; Jemal, A. International Variation in Female Breast Cancer Incidence and Mortality Rates. Cancer Epidemiol. Prev. Biomark. 2015, 24, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Zhou, D.; Zhi, H.; Wang, P.; Gao, Y.; Guo, M.; Yue, M.; Wang, Y.; Shen, W.; et al. Inferences of individual drug responses across diverse cancer types using a novel competing endogenous RNA network. Mol. Oncol. 2018, 12, 1429–1446. [Google Scholar] [CrossRef] [Green Version]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Mullard, A. Parsing clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 447. [Google Scholar] [CrossRef] [PubMed]
- Moreno Roig, E.; Yaromina, A.; Houben, R.; Groot, A.J.; Dubois, L.; Vooijs, M. Prognostic Role of Hypoxia-Inducible Factor-2α Tumor Cell Expression in Cancer Patients: A Meta-Analysis. Front. Oncol. 2018, 8, 224. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef]
- Begley, C.G.; Ellis, L.M. Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Jaroch, K.; Jaroch, A.; Bojko, B. Cell cultures in drug discovery and development: The need of reliable in vitro-in vivo extrapolation for pharmacodynamics and pharmacokinetics assessment. J. Pharm. Biomed. Anal. 2018, 147, 297–312. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. Slas Discov. Adv. Life Sci. R D 2017, 22, 456–472. [Google Scholar] [CrossRef] [Green Version]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Karakashev, S.V.; Reginato, M.J. Progress toward overcoming hypoxia-induced resistance to solid tumor therapy. Cancer Manag. Res. 2015, 7, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, T.J.; van de Velde, C.J.; van Pelt, G.W.; Kroep, J.R.; Julien, J.P.; Smit, V.T.; Tollenaar, R.A.; Mesker, W.E. Prognostic significance of the tumor-stroma ratio: Validation study in node-negative premenopausal breast cancer patients from the EORTC perioperative chemotherapy (POP) trial (10854). Breast Cancer Res. Treat. 2013, 139, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.R.; Wrobel, C.N.; Brugge, J.S. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J. Mammary Gland Biol. Neoplasia 2004, 9, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, C.; Chen, R.; Matsumoto, T.; Schmelzle, T.; Brugge, J.S.; Polverini, P.J.; Mooney, D.J. Engineering tumors with 3D scaffolds. Nat. Methods 2007, 4, 855–860. [Google Scholar] [CrossRef]
- Horning, J.L.; Sahoo, S.K.; Vijayaraghavalu, S.; Dimitrijevic, S.; Vasir, J.K.; Jain, T.K.; Panda, A.K.; Labhasetwar, V. 3-D Tumor Model for In Vitro Evaluation of Anticancer Drugs. Mol. Pharm. 2008, 5, 849–862. [Google Scholar] [CrossRef]
- Shin, C.S.; Kwak, B.; Han, B.; Park, K. Development of an in Vitro 3D Tumor Model to Study Therapeutic Efficiency of an Anticancer Drug. Mol. Pharm. 2013, 10, 2167–2175. [Google Scholar] [CrossRef] [Green Version]
- Brooks, E.A.; Galarza, S.; Gencoglu, M.F.; Cornelison, R.C.; Munson, J.M.; Peyton, S.R. Applicability of drug response metrics for cancer studies using biomaterials. Philos. Trans. R. Soc. B 2019, 374, 20180226. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.A.; Dare, E.V.; Hincke, M. Fibrin: A versatile scaffold for tissue engineering applications. Tissue Eng. Part B Rev. 2008, 14, 199–215. [Google Scholar] [CrossRef]
- Liu, J.; Tan, Y.; Zhang, H.; Zhang, Y.; Xu, P.; Chen, J.; Poh, Y.C.; Tang, K.; Wang, N.; Huang, B. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat. Mater. 2012, 11, 734–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [CrossRef] [PubMed]
- Sidelmann, J.J.; Gram, J.; Jespersen, J.; Kluft, C. Fibrin clot formation and lysis: Basic mechanisms. Semin. Thromb. Hemost. 2000, 26, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; O’Brien, F.J. Understanding the effect of mean pore size on cell activity in collagen-glycosaminoglycan scaffolds. Cell Adhes. Migr. 2010, 4, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Rouwkema, J.; Koopman, B.; Blitterswijk, C.; Dhert, W.; Malda, J. Supply of nutrients to cells in engineered tissues. Biotechnol. Genet. Eng. Rev. 2010, 26, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Niepel, M.; Sorger, P.K. Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Nat. Biotechnol. 2017, 35, 500–502. [Google Scholar] [CrossRef]
- Clark, N.A.; Hafner, M.; Kouril, M.; Williams, E.H.; Muhlich, J.L.; Pilarczyk, M.; Niepel, M.; Sorger, P.K.; Medvedovic, M. GRcalculator: An online tool for calculating and mining dose-response data. BMC Cancer 2017, 17, 698. [Google Scholar] [CrossRef] [Green Version]
- Yates, R.A.; Dowsett, M.; Fisher, G.V.; Selen, A.; Wyld, P.J. Arimidex (ZD1033): A selective, potent inhibitor of aromatase in postmenopausal female volunteers. Br. J. Cancer 1996, 73, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Caan, B.J.; Sweeney, C.; Habel, L.A.; Kwan, M.L.; Kroenke, C.H.; Weltzien, E.K.; Quesenberry, C.P., Jr.; Castillo, A.; Factor, R.E.; Kushi, L.H.; et al. Intrinsic subtypes from the PAM50 gene expression assay in a population-based breast cancer survivor cohort: Prognostication of short- and long-term outcomes. Cancer Epidemiol. Prev. Biomark. 2014, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Prat, A.; Rodriguez-Lescure, A.; Caballero, R.; Ebbert, M.T.; Munarriz, B.; Ruiz-Borrego, M.; Bastien, R.R.; Crespo, C.; Davis, C.; et al. PAM50 proliferation score as a predictor of weekly paclitaxel benefit in breast cancer. Breast Cancer Res. Treat. 2013, 138, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, K.E.; Carey, L.A.; Wazer, D.E. Breast cancer molecular subtypes in patients with locally advanced disease: Impact on prognosis, patterns of recurrence, and response to therapy. Semin. Radiat. Oncol. 2009, 19, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009, 11, R7. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Triulzi, T.; Casalini, P.; Sandri, M.; Ratti, M.; Carcangiu, M.L.; Colombo, M.P.; Balsari, A.; Menard, S.; Orlandi, R.; Tagliabue, E. Neoplastic and stromal cells contribute to an extracellular matrix gene expression profile defining a breast cancer subtype likely to progress. PLoS ONE 2013, 8, e56761. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Breslin, S.; Driscoll, L. The relevance of using 3D cell cultures, in addition to 2D monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 2016, 7, 45745. [Google Scholar] [CrossRef] [Green Version]
- Beekhuijzen, M. The era of 3Rs implementation in developmental and reproductive toxicity (DART) testing: Current overview and future perspectives. Reprod. Toxicol. 2017, 72, 86–96. [Google Scholar] [CrossRef]
- Liu, S.; Yin, N.; Faiola, F. Prospects and Frontiers of Stem Cell Toxicology. Stem Cells Dev. 2017, 26, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, D.D.; Mbemya, G.T.; Bruno, J.B.; Faustino, L.R.; de Figueiredo, J.R.; Rodrigues, A.P.R. In Vitro culture systems as an alternative for female reproductive toxicology studies. Zygote 2019, 27, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.L.; Holzgrefe, H.; Black, L.E.; Brown, M.; Chellman, G.; Copeman, C.; Couch, J.; Creton, S.; Gehen, S.; Hoberman, A.; et al. Pharmaceutical toxicology: Designing studies to reduce animal use, while maximizing human translation. Regul. Toxicol. Pharmacol. 2013, 66, 88–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luz, A.L.; Tokar, E.J. Pluripotent Stem Cells in Developmental Toxicity Testing: A Review of Methodological Advances. Toxicol. Sci. 2018, 165, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef] [Green Version]
- Griffith, L.G.; Swartz, M.A. Capturing complex 3D tissue physiology In Vitro. Nat. Rev. Mol. Cell Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef]
- Cirkel, G.A.; Gadellaa-van Hooijdonk, C.G.; Koudijs, M.J.; Willems, S.M.; Voest, E.E. Tumor heterogeneity and personalized cancer medicine: Are we being outnumbered? Future Oncol. 2014, 10, 417–428. [Google Scholar] [CrossRef]
- Dhiman, H.K.; Ray, A.R.; Panda, A.K. Three-dimensional chitosan scaffold-based MCF-7 cell culture for the determination of the cytotoxicity of tamoxifen. Biomaterials 2005, 26, 979–986. [Google Scholar] [CrossRef]
- Park, K.M.; Gerecht, S. Hypoxia-inducible hydrogels. Nat. Commun. 2014, 5, 4075. [Google Scholar] [CrossRef] [Green Version]
- Loessner, D.; Stok, K.S.; Lutolf, M.P.; Hutmacher, D.W.; Clements, J.A.; Rizzi, S.C. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials 2010, 31, 8494–8506. [Google Scholar] [CrossRef] [Green Version]
- De la Puente, P.; Ludena, D.; Fernandez, A.; Aranda, J.L.; Varela, G.; Iglesias, J. Autologous fibrin scaffolds cultured dermal fibroblasts and enriched with encapsulated bFGF for tissue engineering. J. Biomed. Mater. Res. Part A 2011, 99, 648–654. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Ludena, D.; Lopez, M.; Ramos, J.; Iglesias, J. Differentiation within autologous fibrin scaffolds of porcine dermal cells with the mesenchymal stem cell phenotype. Exp. Cell Res. 2013, 319, 144–152. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Ludena, D. Cell culture in autologous fibrin scaffolds for applications in tissue engineering. Exp. Cell Res. 2014, 322, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Germain, L.; De Berdt, P.; Vanacker, J.; Leprince, J.; Diogenes, A.; Jacobs, D.; Vandermeulen, G.; Bouzin, C.; Preat, V.; Dupont-Gillain, C.; et al. Fibrin hydrogels to deliver dental stem cells of the apical papilla for regenerative medicine. Regen. Med. 2015, 10, 153–167. [Google Scholar] [CrossRef]
- Clark, R.A.F. Fibrin Is a Many Splendored Thing. J. Investig. Dermatol. 2003, 121. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Calar, K.; Evans, C.; Petrasko, M.; de la Puente, P. Bioengineering a novel 3D in-vitro model to recreate physiological oxygen levels and tumor-immune interactions. Biorxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Thippabhotla, S.; Zhong, C.; He, M. 3D cell culture stimulates the secretion of In Vivo like extracellular vesicles. Sci. Rep. 2019, 9, 13012. [Google Scholar] [CrossRef] [Green Version]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Watkins, G.; Parr, C.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. Stromal cell derived factor-1: Its influence on invasiveness and migration of breast cancer cells in vitro, and its association with prognosis and survival in human breast cancer. Breast Cancer Res. 2005, 7, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Hojilla, C.V.; Wood, G.A.; Khokha, R. Inflammation and breast cancer. Metalloproteinases as common effectors of inflammation and extracellular matrix breakdown in breast cancer. Breast Cancer Res. 2008, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef]
- Friedman, S.L. Cytokines and fibrogenesis. Semin. Liver Dis. 1999, 19, 129–140. [Google Scholar] [CrossRef]
- Bränn, E.; Edvinsson, Å.; Rostedt Punga, A.; Sundström-Poromaa, I.; Skalkidou, A. Inflammatory and anti-inflammatory markers in plasma: From late pregnancy to early postpartum. Sci. Rep. 2019, 9, 1863. [Google Scholar] [CrossRef]
- Norum, L.F.; Erikstein, B.; Nustad, K. Elevated CA125 in breast cancer—A sign of advanced disease. Tumour Biol. 2001, 22, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Cao, Y.; Liu, X.; Zeng, X.-T.; Li, Y. Serum CA125 is a predictive marker for breast cancer outcomes and correlates with molecular subtypes. Oncotarget 2017, 8, 63963–63970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.; Liu, H.; Sun, X.; Li, X.; Zhao, S.; Ma, R. Plasma fibrinogen level may be a possible marker for the clinical response and prognosis of patients with breast cancer receiving neoadjuvant chemotherapy. Tumor Biol. 2017, 39, 1010428317700002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shida, N.; Kurasawa, R.; Maki, Y.; Toyama, Y.; Dobashi, T.; Yamamoto, T. Study of plasma coagulation induced by contact with calcium chloride solution. Soft Matter 2016, 12, 9471–9476. [Google Scholar] [CrossRef] [PubMed]
- Siebenlist, K.R.; Meh, D.A.; Mosesson, M.W. Protransglutaminase (factor XIII) mediated crosslinking of fibrinogen and fibrin. Thromb. Haemost. 2001, 86, 1221–1228. [Google Scholar]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naghieh, S.; Karamooz-Ravari, M.R.; Sarker, M.D.; Karki, E.; Chen, X. Influence of crosslinking on the mechanical behavior of 3D printed alginate scaffolds: Experimental and numerical approaches. J. Mech. Behav. Biomed. Mater. 2018, 80, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bou-Akl, T.; Banglmaier, R.; Miller, R.; VandeVord, P. Effect of crosslinking on the mechanical properties of mineralized and non-mineralized collagen fibers. J. Biomed. Mater. Res. Part A 2013, 101, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- De la Puente, P.; Quan, N.; Hoo, R.S.; Muz, B.; Gilson, R.C.; Luderer, M.; King, J.; Achilefu, S.; Salama, N.N.; Vij, R.; et al. Newly established myeloma-derived stromal cell line MSP-1 supports multiple myeloma proliferation, migration, and adhesion and induces drug resistance more than normal-derived stroma. Haematologica 2016, 101, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Gargotti, M.; Lopez-Gonzalez, U.; Byrne, H.J.; Casey, A. Comparative studies of cellular viability levels on 2D and 3D in vitro culture matrices. Cytotechnology 2018, 70, 261–273. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Asselin, E.; Parent, S.; Sykes, P.H.; Evans, J.J. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp. Cell Res. 2013, 319, 75–87. [Google Scholar] [CrossRef]
- Pineda, E.T.; Nerem, R.M.; Ahsan, T. Differentiation patterns of embryonic stem cells in two-versus three-dimensional culture. Cells Tissues Organs 2013, 197, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, J.; Inoue, M. Application of Cancer Organoid Model for Drug Screening and Personalized Therapy. Cells 2019, 8, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, B.; Song, X.; Li, N.; Zhao, D.; Li, M.; Zhao, Z. Breast cancer organoids from a patient with giant papillary carcinoma as a high-fidelity model. Cancer Cell Int. 2020, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Mahecha, A.; Lafleur, J.; Pelmus, M.; Seguin, C.; Lan, C.; Discepola, F.; Kovacina, B.; Christodoulopoulos, R.; Salvucci, O.; Mihalcioiu, C.; et al. The identification of challenges in tissue collection for biomarker studies: The Q-CROC-03 neoadjuvant breast cancer translational trial experience. Mod. Pathol. 2017, 30, 1567–1576. [Google Scholar] [CrossRef]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Niepel, M.; Hafner, M.; Mills, C.E.; Subramanian, K.; Williams, E.H.; Chung, M.; Gaudio, B.; Barrette, A.M.; Stern, A.D.; Hu, B.; et al. A Multi-center Study on the Reproducibility of Drug-Response Assays in Mammalian Cell Lines. Cell Syst. 2019, 9, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Witte, C.J.; Espejo Valle-Inclan, J.; Hami, N.; Lohmussaar, K.; Kopper, O.; Vreuls, C.P.H.; Jonges, T.N.; van Diest, P.J.; Nguyen, L.; Clevers, H.; et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 2020, 31, 107762. [Google Scholar] [CrossRef]
- Hongisto, V.; Jernström, S.; Fey, V.; Mpindi, J.-P.; Kleivi Sahlberg, K.; Kallioniemi, O.; Perälä, M. High-Throughput 3D Screening Reveals Differences in Drug Sensitivities between Culture Models of JIMT1 Breast Cancer Cells. PLoS ONE 2013, 8, e77232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, P.; Gupta, M. Personalized & Precision Medicine in Cancer: A Theranostic Approach. Curr. Radiopharm. 2017, 10, 166–170. [Google Scholar] [CrossRef]
- Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Beck, A.H.; Aerts, H.J.W.L.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2013, 504, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini Bereshneh, A.; Morshedi, F.; Hematyar, M.; Kaki, A.; Garshasbi, M. Pharmacogenetics and Personalized Medicine in Pancreatic Cancer. Acta Med. Iran. 2017, 55, 194–199. [Google Scholar]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2016, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Giacomotto, J.; Segalat, L. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 2010, 160, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.-H.; Smith, A.S.T.; Lone, S.; Kwon, S.; Kim, D.-H. Biomimetic 3D Tissue Models for Advanced High-Throughput Drug Screening. J. Lab. Autom. 2015, 20, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int. J. Mol. Sci. 2012, 13, 427–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curigliano, G.; Criscitiello, C. Successes and limitations of targeted cancer therapy in breast cancer. Prog. Tumor Res. 2014, 41, 15–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, V.; Pagès, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.L.; Hermans, J.; McDonagh, J.; McDonagh, R.P.; Carr, M. Effects of calcium ion and covalent crosslinking on formation and elasticity of fibrin cells. Thromb. Res. 1975, 6, 255–265. [Google Scholar] [CrossRef]
- Stang, L.J.; Mitchell, L.G. Fibrinogen. Methods Mol. Biol. 2013, 992, 181–192. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Zylla, J.S.; Bhattacharya, S.; Calar, K.; Hartman, T.W.; Bhardwaj, R.D.; Miskimins, W.K.; de la Puente, P.; Gnimpieba, E.Z.; Messerli, S.M. Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers 2020, 12, 756. [Google Scholar] [CrossRef] [Green Version]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Doubling Time | Origin | Pathology | Subtype | ER | PR | HER2 |
|---|---|---|---|---|---|---|---|
| MDA-MB-231 | 38 h | Pleural Effusion | Adenocarcinoma | Triple Negative B | − | − | − |
| MDA-MB-453 | 38 h | Pleural Effusion | Adenocarcinoma | HER2 Positive | − | − | + |
| ZR-75-1 | 54 h | Ascites | Invasive Ductal Carcinoma | Luminal A | + | −/+ | − |
| MCF7 | 48 h | Pleural Effusion | Invasive Ductal Carcinoma | Luminal A | + | + | − |
| SK-BR-3 | 30 h | Pleural Effusion | Adenocarcinoma | HER2 Positive | − | − | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calar, K.; Plesselova, S.; Bhattacharya, S.; Jorgensen, M.; de la Puente, P. Human Plasma-Derived 3D Cultures Model Breast Cancer Treatment Responses and Predict Clinically Effective Drug Treatment Concentrations. Cancers 2020, 12, 1722. https://doi.org/10.3390/cancers12071722
Calar K, Plesselova S, Bhattacharya S, Jorgensen M, de la Puente P. Human Plasma-Derived 3D Cultures Model Breast Cancer Treatment Responses and Predict Clinically Effective Drug Treatment Concentrations. Cancers. 2020; 12(7):1722. https://doi.org/10.3390/cancers12071722
Chicago/Turabian StyleCalar, Kristin, Simona Plesselova, Somshuvra Bhattacharya, Megan Jorgensen, and Pilar de la Puente. 2020. "Human Plasma-Derived 3D Cultures Model Breast Cancer Treatment Responses and Predict Clinically Effective Drug Treatment Concentrations" Cancers 12, no. 7: 1722. https://doi.org/10.3390/cancers12071722
APA StyleCalar, K., Plesselova, S., Bhattacharya, S., Jorgensen, M., & de la Puente, P. (2020). Human Plasma-Derived 3D Cultures Model Breast Cancer Treatment Responses and Predict Clinically Effective Drug Treatment Concentrations. Cancers, 12(7), 1722. https://doi.org/10.3390/cancers12071722