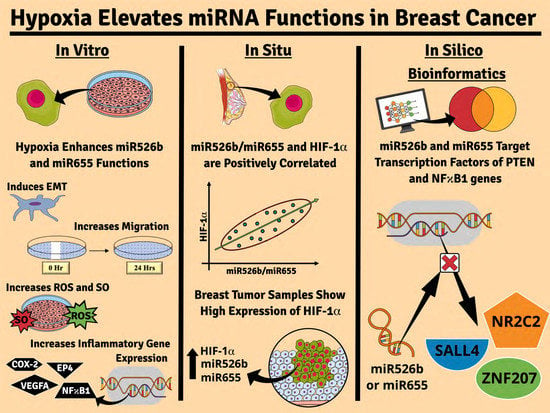
Chemically Induced Hypoxia Enhances miRNA Functions in Breast Cancer

 , ,
, , 
Abstract
:
1. Introduction
2. Results
2.1. HIF-1α Gene and Protein Expression in Normoxia
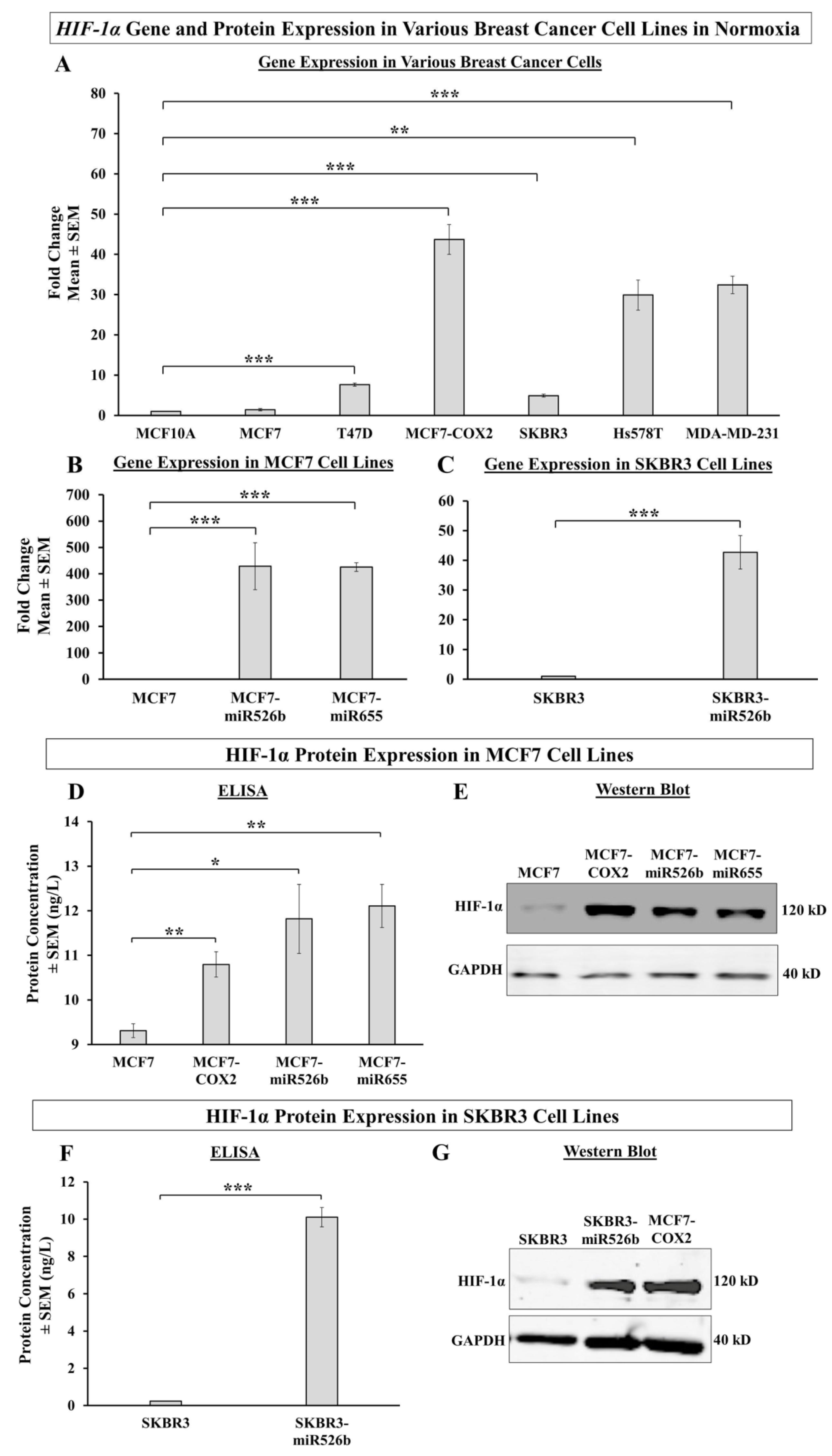
2.1.1. HIF-1α Gene Expression in Various Breast Cancer Cell Lines
2.1.2. HIF-1α Protein Expression in Various Breast Cancer Cell Lines
2.2. Induction of Hypoxia Using CoCl2
2.2.1. HIF-1α Gene Expression in Hypoxia
2.2.2. HIF-1α Protein Expression in Hypoxia
2.2.3. Analysis of VHL Gene Expression in Hypoxia
2.2.4. Hypoxia Enhances miRNA Expression
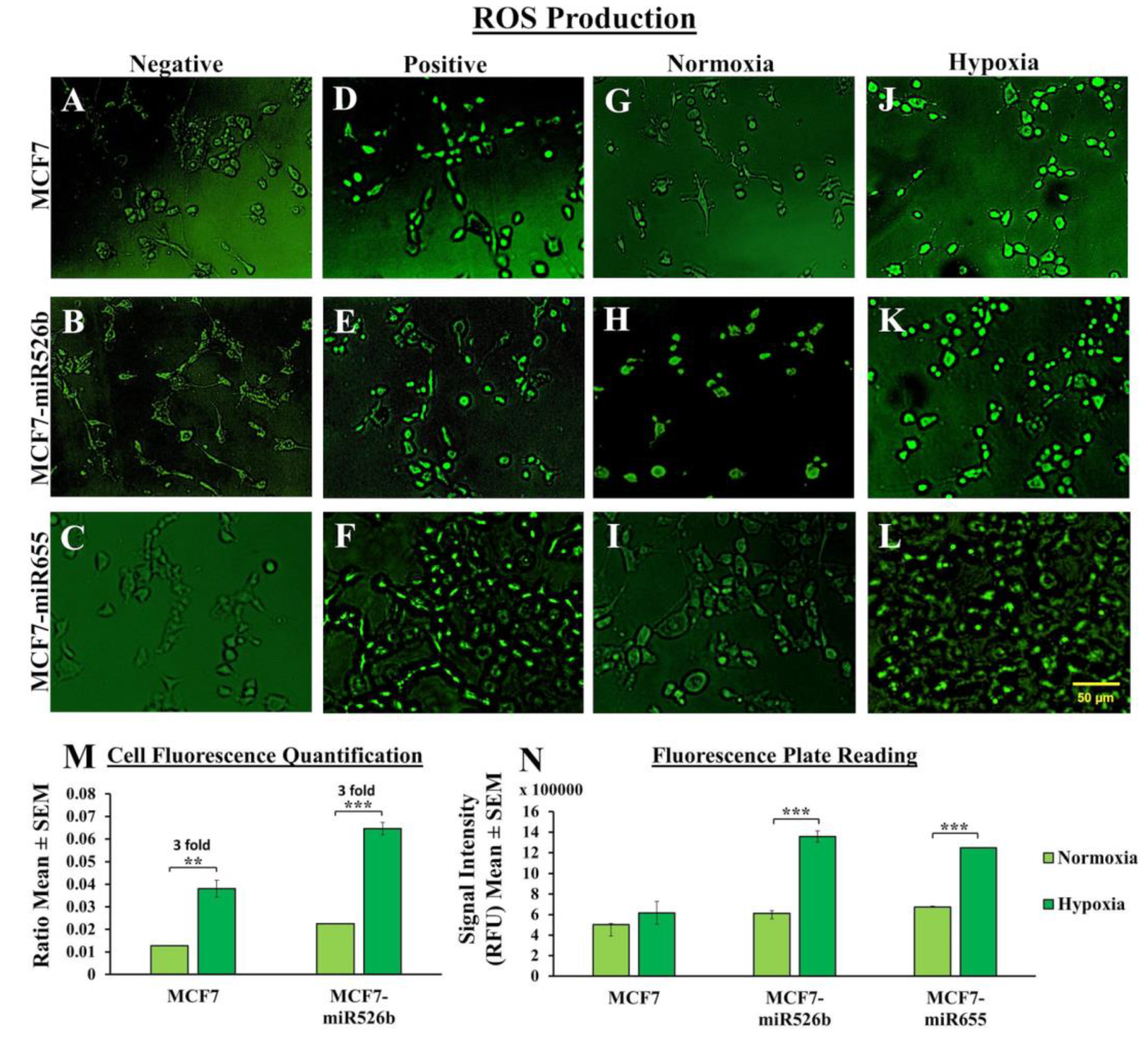
2.3. Hypoxia Induces Oxidative Stress
2.3.1. Fluorescence Microscopy Assay to Measure Cellular Fluorescence
2.3.2. Fluorescence Microplate Assay to Measure Total Fluorescence
2.3.3. Overexpression of TXNRD1
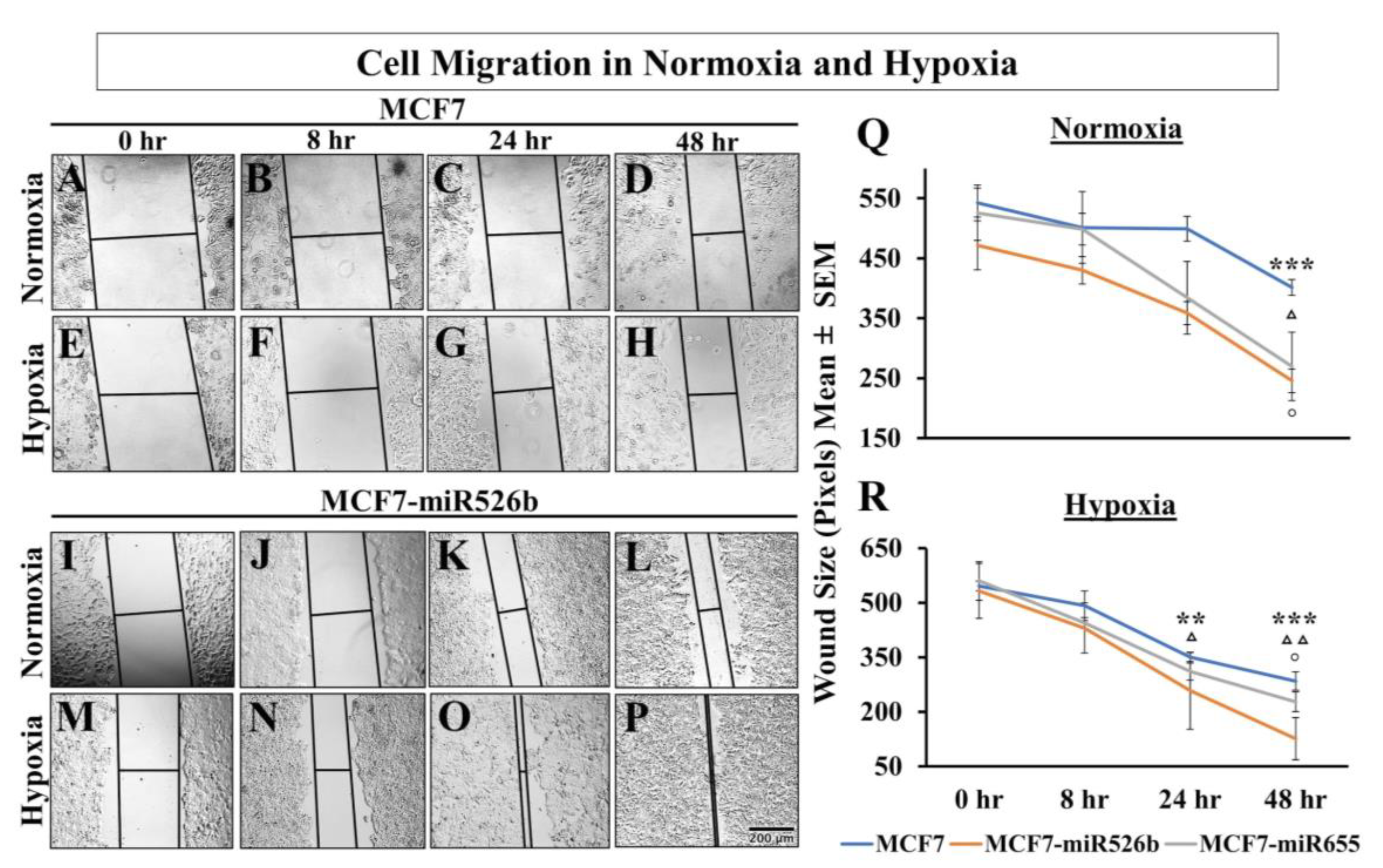
2.4. Hypoxia Promotes EMT in miRNA-High Cells
2.4.1. Hypoxic Condition Regulates EMT Markers Expression in Cancer Cells
2.4.2. Hypoxic Condition Promotes Migration of miRNA-High Cells
2.5. Hypoxia Promotes Inflammatory Gene Expression and Vascular Mimicry in miRNA-High Cells
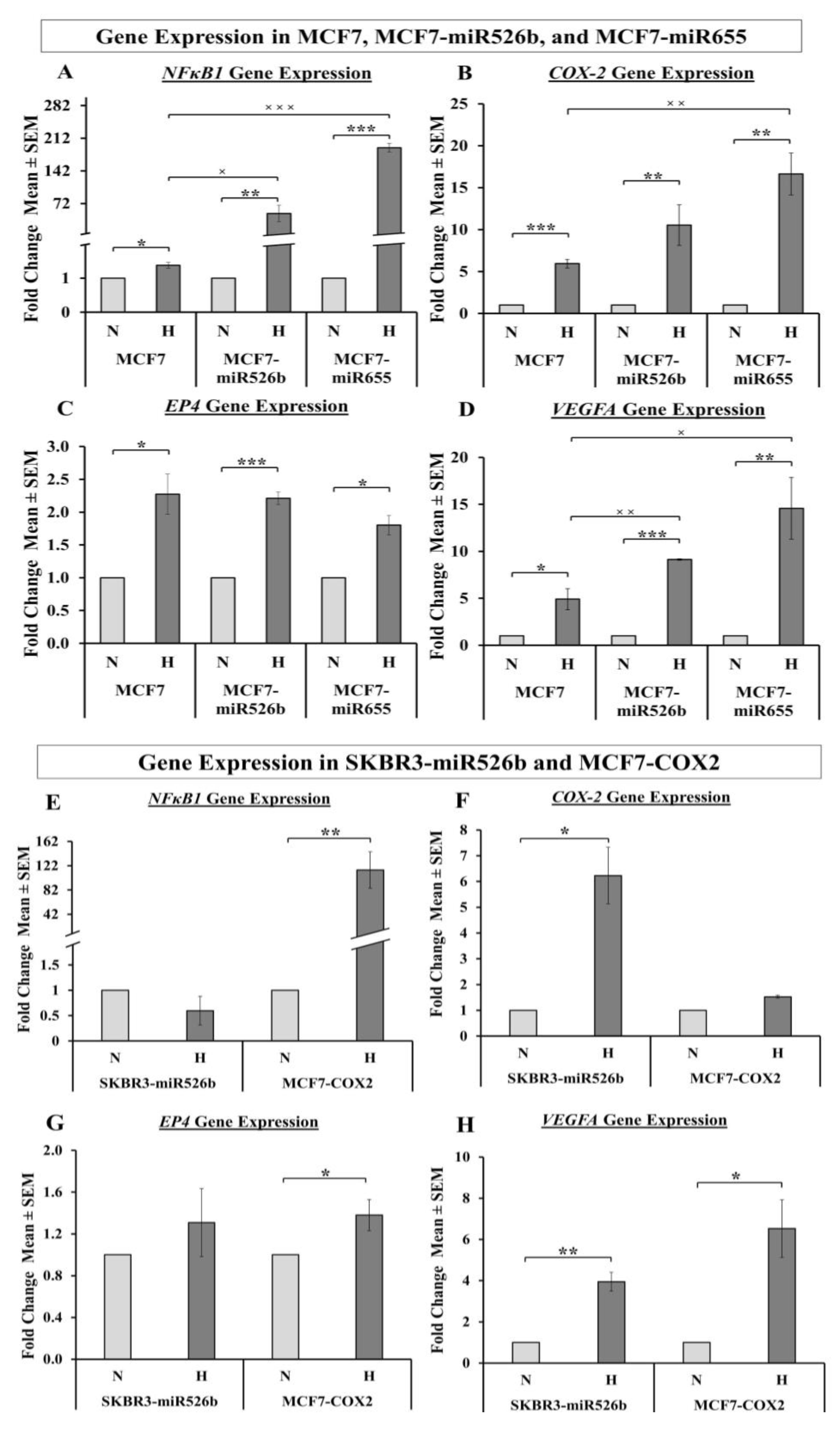
2.5.1. Hypoxia Promotes Expression of NFκB1, COX-2, and EP4
2.5.2. Hypoxia Promotes VEGFA Gene Expression
2.5.3. Hypoxia Promotes Vascular Mimicry
2.6. Inhibition of Hypoxia-Enhanced Functions in miRNA-High Cells
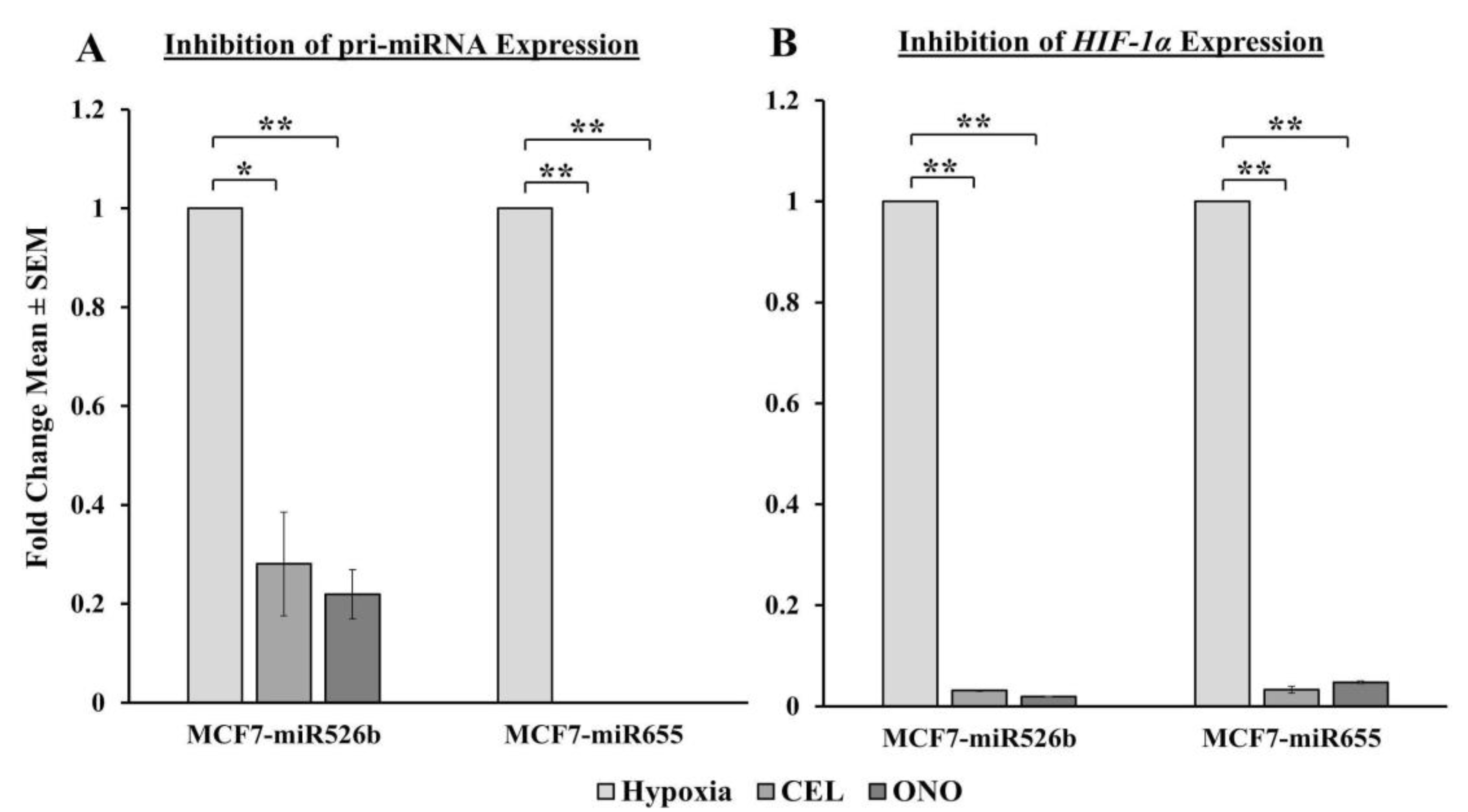
2.6.1. Pri-miRNA and HIF-1α Gene Expression Abrogated with COX-2 Inhibitor and EP4 Antagonist
2.6.2. Inhibition of ROS/SO Production
2.6.3. Inhibition of Cell Migration
2.6.4. Inhibition of Vascular Mimicry
2.7. Linking COX-2, EP4, and PI3K/Akt Pathways with Hypoxia and miRNAs
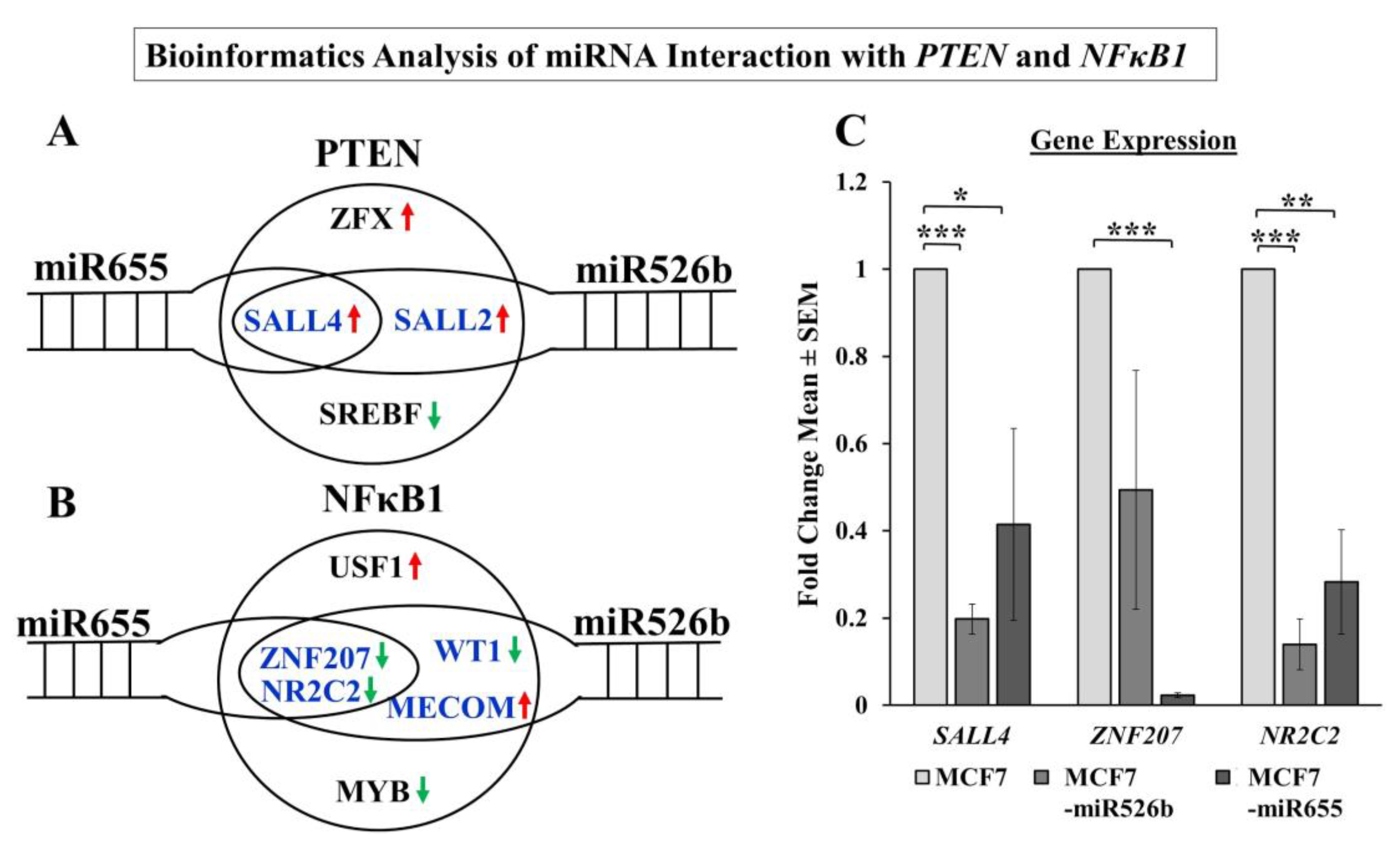
2.8. Bioinformatics Analysis and Regulation of PTEN and NFκB1 by miRNA
2.8.1. PTEN Regulation
2.8.2. NFκB1 Regulation
2.9. miR526b and miR655 Expression Significantly Correlates with HIF-1α Expression in Human Breast Tumors
2.9.1. Ontario Tumor Bank Sample Demography
2.9.2. HIF-1α and miRNA Expression in Breast Tumor and Control Tissues
2.9.3. Data Extracted from the cBioPortal Database Via TCGA
3. Discussion
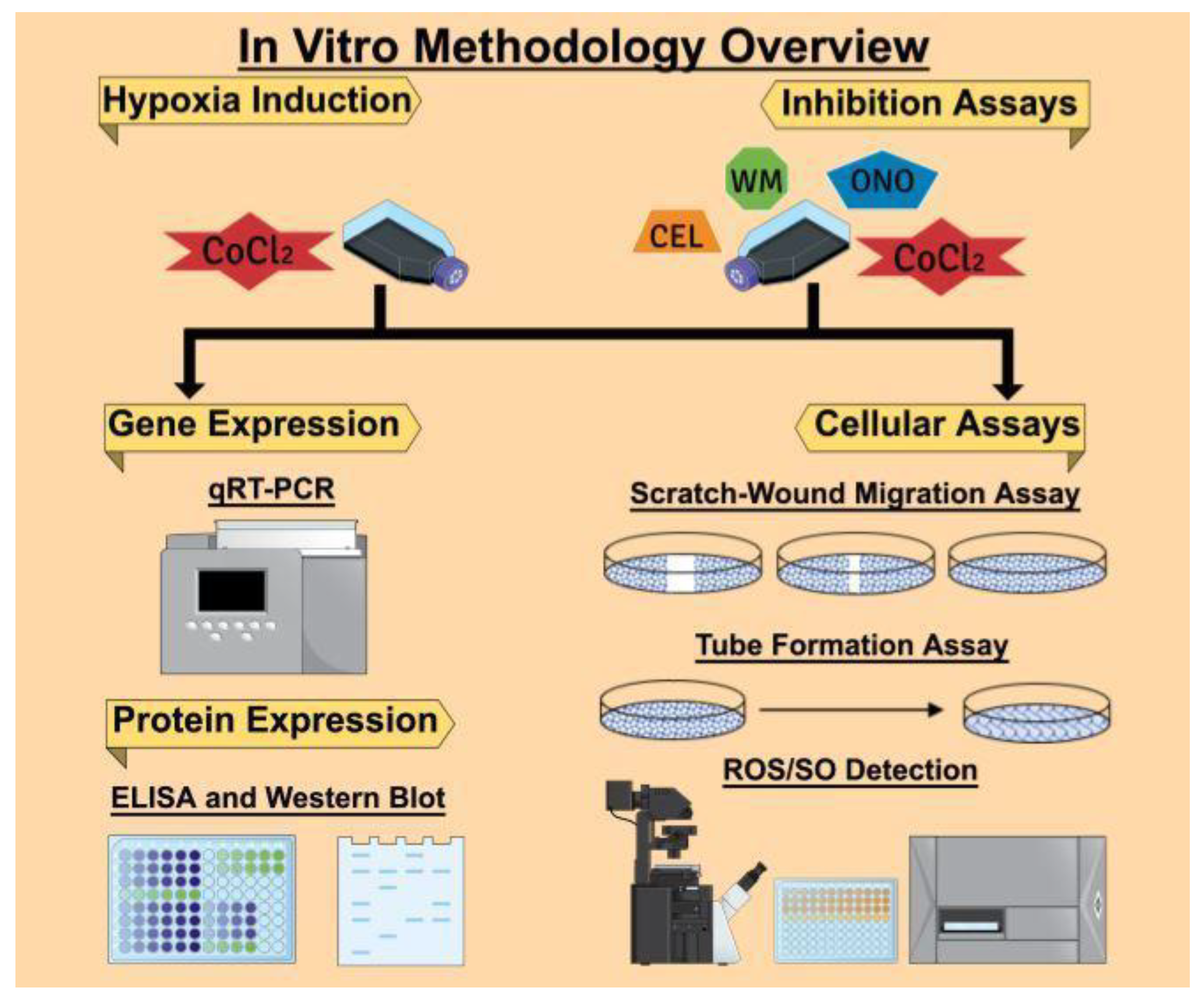
4. Materials and Methods
4.1. Ethics Statements
4.2. Cell Culture
4.3. Drugs and Chemicals
4.4. Hypoxia Induction In Vitro with CoCl2 Treatment
4.5. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
4.6. Enzyme-Linked Immunosorbent Assay (ELISA) Analysis of HIF-1α
4.7. Western Blot Analysis
4.8. Fluorescence Microplate Assay
4.9. Fluorescence Microscopy Assay
4.10. Scratch-Wound Migration Assay
4.11. Tube Formation Assay
4.12. Bioinformatics Analysis
4.13. Human Breast Cancer Tissue Samples
4.14. In Silico Analysis of cBioPortal Data via TCGA
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CEL | Celecoxib |
| CDH1 | E-cadherin (Epithelial cadherin) |
| CoCl2 | Cobalt chloride |
| COX-2 | Cyclooxygenase-2 |
| CPEB2 | Cytoplasmic polyadenylation element-binding protein 2 |
| ELISA | Enzyme-linked immunosorbent assay |
| EMT | Epithelial-to-mesenchymal transition |
| EP4 | Prostaglandin E2 receptor 4 |
| ER | Estrogen receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| miRNA/miR | MicroRNA |
| mRNA | Messenger RNA |
| NFκB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| NFκB1 | Nuclear factor NF-kappa-B p105 subunit |
| NR2C2 | Testicular receptor 4 |
| ONO | ONO-AE3-208 |
| PGE2 | Prostaglandin E₂ |
| PHD | Prolyl hydroxylase domain |
| PI3K | Phosphoinositide 3-kinases |
| PR | Progesterone receptor |
| Pri-miRNA | Primary miRNA |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SALL2 | Sal-like protein 2 |
| SALL4 | Sal-like protein 4 |
| SLC | Stem-like cell |
| SNAIL | Zinc finger protein SNAI1 |
| SO | Superoxide |
| TCGA | The Cancer Genome Atlas |
| TWIST1 | TWIST1-related protein 1 |
| TXNRD1 | Thioredoxin reductase 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFA | Vascular endothelial growth factor A |
| VHL | Von Hippel–Lindau tumor suppressor |
| VIM | Vimentin |
| WM | Wortmannin |
| ZNF207 | Zinc finger protein 207 |
References
- Canadian Cancer Statistics 2019; Canadian Cancer Society: Toronto, ON, Canada, 2019.
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, S.; Nault, B.; Ugwuagbo, K.C.; Maiti, S.; Majumder, M. Mir526b and Mir655 Promote Tumour Associated Angiogenesis and Lymphangiogenesis in Breast Cancer. Cancers 2019, 11, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, G.-O.; Seita, J.; Hong, B.-J.; Kim, Y.-E.; Bok, S.; Lee, C.-J.; Kim, K.S.; Lee, J.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc. Natl. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Jin, X.; Gao, Y.; Yuan, H.; Wang, F.; Cao, X. DZNep inhibits Hif-1α and Wnt signalling molecules to attenuate the proliferation and invasion of BGC-823 gastric cancer cells. Oncol. Lett. 2019, 18, 4308–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengellur, A.; Woods, B.G.; Ryan, H.E.; Johnson, R.S.; Lapres, J.J. Gene Expression Profiling of the Hypoxia Signaling Pathway in Hypoxia-Inducible Factor 1α Null Mouse Embryonic Fibroblasts. Gene Expr. 2003, 11, 181–197. [Google Scholar] [CrossRef]
- Camps, C.; Saini, H.; Mole, D.R.; Choudhry, H.; Reczko, M.; Guerra-Assunção, J.A.; Tian, Y.-M.; Buffa, F.M.; Harris, A.L.; Hatzigeorgiou, A.; et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol. Cancer 2014, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, M.; Layng, F.I.A.L.; Lemos, R.; Garcia, G.; James, B.P.; Sevilla, M.; Soldevilla, F.; Baaten, B.J.; De Jong, P.R.; Koh, M.Y.; et al. Absence of HIF1A Leads to Glycogen Accumulation and an Inflammatory Response That Enables Pancreatic Tumor Growth. Cancer Res. 2019, 79, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Del Rey, M.J.; Valín, Á.; Usategui, A.; García-Herrero, C.M.; Sánchez-Aragó, M.; Cuezva, J.M.; Galindo, M.; Bravo, B.; Cañete, J.D.; Blanco, F.J.; et al. Hif-1α Knockdown Reduces Glycolytic Metabolism and Induces Cell Death of Human Synovial Fibroblasts Under Normoxic Conditions. Sci. Rep. 2017, 7, 3644. [Google Scholar] [CrossRef] [Green Version]
- Loh, H.-Y.; Norman, B.P.; Lai, K.-S.; Rahman, N.M.A.N.A.; Alitheen, N.; Osman, M.A. The Regulatory Role of MicroRNAs in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 4940. [Google Scholar] [CrossRef] [Green Version]
- Krutilina, R.; Sun, W.; Sethuraman, A.; Brown, M.; Seagroves, T.N.; Pfeffer, L.M.; Ignatova, T.; Fan, M. MicroRNA-18a inhibits hypoxia-inducible factor 1α activity and lung metastasis in basal breast cancers. Breast Cancer Res. 2014, 16, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Landman, E.; Liu, L.; Hess, D.; Lala, P.K. COX-2 Elevates Oncogenic miR-526b in Breast Cancer by EP4 Activation. Mol. Cancer Res. 2015, 13, 1022–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Dunn, L.; Liu, L.; Hasan, A.; Vincent, K.; Brackstone, M.; Hess, D.; Lala, P.K. COX-2 induces oncogenic micro RNA miR655 in human breast cancer. Sci. Rep. 2018, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Tordjman, J.; Majumder, M.; Amiri, M.; Hasan, A.; Hess, D.A.; Lala, P.K. Tumor suppressor role of cytoplasmic polyadenylation element binding protein 2 (CPEB2) in human mammary epithelial cells. BMC Cancer 2019, 19, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Bock, C.; Paulsen, M. Structural conservation versus functional divergence of maternally expressed microRNAs in the Dlk1/Gtl2 imprinting region. BMC Genom. 2008, 9, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braschi, B.; Denny, P.; Gray, K.; Jones, T.E.M.; Seal, R.; Tweedie, S.; Yates, B.; Bruford, E. Genenames.org: The HGNC and VGNC resources in 2019. Nucleic Acids Res. 2018, 47, D786–D792. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Flores, E.R.; Brohl, A.S. Chromosome 19 miRNA cluster and CEBPB expression specifically mark and potentially drive triple negative breast cancers. PLoS ONE 2018, 13, e0206008. [Google Scholar] [CrossRef] [Green Version]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Yang, F.; Shao, C.; Wei, K.; Jing, X.; Qin, Z.; Shi, Y.; Shu, Y.; Shen, H. miR-942 promotes tumor migration, invasion, and angiogenesis by regulating EMT via BARX2 in non-small-cell lung cancer. J. Cell. Physiol. 2019, 234, 23596–23607. [Google Scholar] [CrossRef]
- Azad, T.; Ghahremani, M.; Yang, X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells 2019, 8, 407. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.; Seftor, E.A.; Kirschmann, D.A.; Seftor, R.E. Molecular biology of breast cancer metastasis Molecular expression of vascular markers by aggressive breast cancer cells. Breast Cancer Res. 2000, 2, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaranta, V. Cell Migration through Extracellular Matrix. J. Cell Biol. 2000, 149, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nantajit, D.; Lin, N.; Li, J.J. The network of epithelial-mesenchymal transition: Potential new targets for tumor resistance. J. Cancer Res. Clin. Oncol. 2014, 141, 1697–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buetler, T.M.; Krauskopf, A.; Rüegg, U. Role of Superoxide as a Signaling Molecule. Physiology 2004, 19, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Feser, R.; Nault, B.; Hunter, S.; Maiti, S.; Ugwuagbo, K.C.; Majumder, M. miR526b and miR655 Induce Oxidative Stress in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 4039. [Google Scholar] [CrossRef] [Green Version]
- Piret, J.-P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a Chemical Inducer of Hypoxia-Inducible Factor-1, and Hypoxia Reduce Apoptotic Cell Death in Hepatoma Cell Line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef]
- Li, Q.; Ma, R.; Zhang, M. CoCl2 increases the expression of hypoxic markers HIF-1α, VEGF and CXCR4 in breast cancer MCF-7 cells. Oncol. Lett. 2017, 15, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Rana, N.K.; Singh, P.; Koch, B. CoCl2 simulated hypoxia induce cell proliferation and alter the expression pattern of hypoxia associated genes involved in angiogenesis and apoptosis. Biol. Res. 2019, 52, 12. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Yotnda, P. Induction and Testing of Hypoxia in Cell Culture. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [Green Version]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.-M.; Hess, D.; Lala, P.K. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef] [Green Version]
- Xin, X.; Majumder, M.; Girish, G.V.; Mohindra, V.; Maruyama, T.; Lala, P.K. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Investig. 2012, 92, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, P.; Girish, G.V.; Majumder, M.; Xin, X.; Tutunea-Fatan, E.; Lala, P.K. PGE2 promotes breast cancer-associated lymphangiogenesis by activation of EP4 receptor on lymphatic endothelial cells. BMC Cancer 2017, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Nandi, P.; Omar, A.; Ugwuagbo, K.C.; Lala, P.K. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Skanderup, A.J.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Bandara, K.V.; Michael, M.Z.; Gleadle, J.M. MicroRNA Biogenesis in Hypoxia. MicroRNA 2017, 6, 80–96. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Kim, C.W.; Oh, E.-T.; Kim, J.M.; Park, J.-S.; Lee, D.H.; Lee, J.-S.; Kim, K.K.; Park, H.J. Hypoxia-induced microRNA-590-5p promotes colorectal cancer progression by modulating matrix metalloproteinase activity. Cancer Lett. 2017, 416, 31–41. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Yagi, S.; Ito, T.; Lowenstein, C.J. MicroRNA-22 Regulates Hypoxia Signaling in Colon Cancer Cells. PLoS ONE 2011, 6, e20291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.-G.; Croce, C.M.; Negrini, M.; et al. A MicroRNA Signature of Hypoxia. Mol. Cell. Biol. 2006, 27, 1859–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free. Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.-Z.; Jin, M.-Z.; Dai, J.-X.; Sun, W.; Feng, J.-H.; Jin, W.-L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef] [Green Version]
- Ugwuagbo, K.C.; Maiti, S.; Omar, A.; Hunter, S.; Nault, B.; Northam, C.; Majumder, M. Prostaglandin E2 promotes embryonic vascular development and maturation in zebrafish. Biol. Open 2019, 8, bio039768. [Google Scholar] [CrossRef] [Green Version]
- Majumder, M.; Xin, X.; Liu, L.; Girish, G.V.; Lala, P.K. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014, 105, 1142–1151. [Google Scholar] [CrossRef]
- Sun, G.; Zhou, Y.; Li, H.; Guo, Y.; Shan, J.; Xia, M.; Li, Y.; Li, S.; Long, D.; Li, Y. Over-expression of microRNA-494 up-regulates hypoxia-inducible factor-1 alpha expression via PI3K/Akt pathway and protects against hypoxia-induced apoptosis. J. Biomed. Sci. 2013, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Dimitrov, T.; Yong, K.J.; Tatetsu, H.; Jeong, H.-W.; Luo, H.R.; Bradner, J.E.; Tenen, D.G.; Chai, L. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 2013, 121, 1413–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Lin, C.; Wang, X.; Peng, X.; Li, Y.; Wang, M.; Zhao, Z.; Wu, X.; Shi, D.; Xiao, Y.; et al. Epigenetic silencing of SALL2 confers tamoxifen resistance in breast cancer. EMBO Mol. Med. 2019, 11, e10638. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Tang, L.; Dong, H.; Dong, Z.; Zhang, L.; Fu, J.; Su, X.; Zhang, T.; Fu, H.; Han, L.; et al. Oncogenic HER2 fusions in gastric cancer. J. Transl. Med. 2015, 13, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyr, C.-R.; Hu, Y.-C.; Kim, E.; Chang, C. Modulation of Estrogen Receptor-mediated Transactivation by Orphan Receptor TR4 in MCF-7 Cells. J. Biol. Chem. 2002, 277, 14622–14628. [Google Scholar] [CrossRef] [Green Version]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’Ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, B. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Benign N = 20 (%) | Malignant N = 96 (%) | |
|---|---|---|---|
| Sex | Female | 20 (100) | 93 (96.88) |
| Male | 0 (0) | 3 (3.13) | |
| Age Distribution (Years) | Range | 52–87 | 27–92 |
| Age (years) | Mean ± SD | 66 ± 11 | 63 ± 17 |
| Smoking | Smokers | 1 (5) | 24 (25) |
| Pack Year (PY) ± SD | 40 | 27 ± 19 | |
| Alcohol Consumption | Social or Occasional Drinker | 5 (25) | 28 (29.17) |
| Regular Drinker | 0 (0) | 3 (3.13) | |
| ER Status | Positive | N/A | 37 (38.58) |
| Negative | N/A | 19 (19.79) | |
| Unknown | N/A | 6 (6.25) | |
| PR Status | Positive | N/A | 31 (32.29) |
| Negative | N/A | 30 (31.25) | |
| Unknown | N/A | 6 (6.25) | |
| HER2 Status | Positive | N/A | 21 (21.88) |
| Negative | N/A | 61 (63.54) | |
| Unknown | N/A | 14 (14.58) | |
| ER, PR, HER2 Status | Negative | N/A | 10 (10.42) |
| Tumor Stage | |||
| I * | N/A | 7 (7.29) | |
| II | N/A | 45 (46.87) | |
| III | N/A | 39 (40.63) | |
| IV | N/A | 5 (5.21) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gervin, E.; Shin, B.; Opperman, R.; Cullen, M.; Feser, R.; Maiti, S.; Majumder, M. Chemically Induced Hypoxia Enhances miRNA Functions in Breast Cancer. Cancers 2020, 12, 2008. https://doi.org/10.3390/cancers12082008
Gervin E, Shin B, Opperman R, Cullen M, Feser R, Maiti S, Majumder M. Chemically Induced Hypoxia Enhances miRNA Functions in Breast Cancer. Cancers. 2020; 12(8):2008. https://doi.org/10.3390/cancers12082008
Chicago/Turabian StyleGervin, Emma, Bonita Shin, Reid Opperman, Mackenzie Cullen, Riley Feser, Sujit Maiti, and Mousumi Majumder. 2020. "Chemically Induced Hypoxia Enhances miRNA Functions in Breast Cancer" Cancers 12, no. 8: 2008. https://doi.org/10.3390/cancers12082008
APA StyleGervin, E., Shin, B., Opperman, R., Cullen, M., Feser, R., Maiti, S., & Majumder, M. (2020). Chemically Induced Hypoxia Enhances miRNA Functions in Breast Cancer. Cancers, 12(8), 2008. https://doi.org/10.3390/cancers12082008