Telomeres and Telomerase in the Development of Liver Cancer



Abstract
:1. Introduction
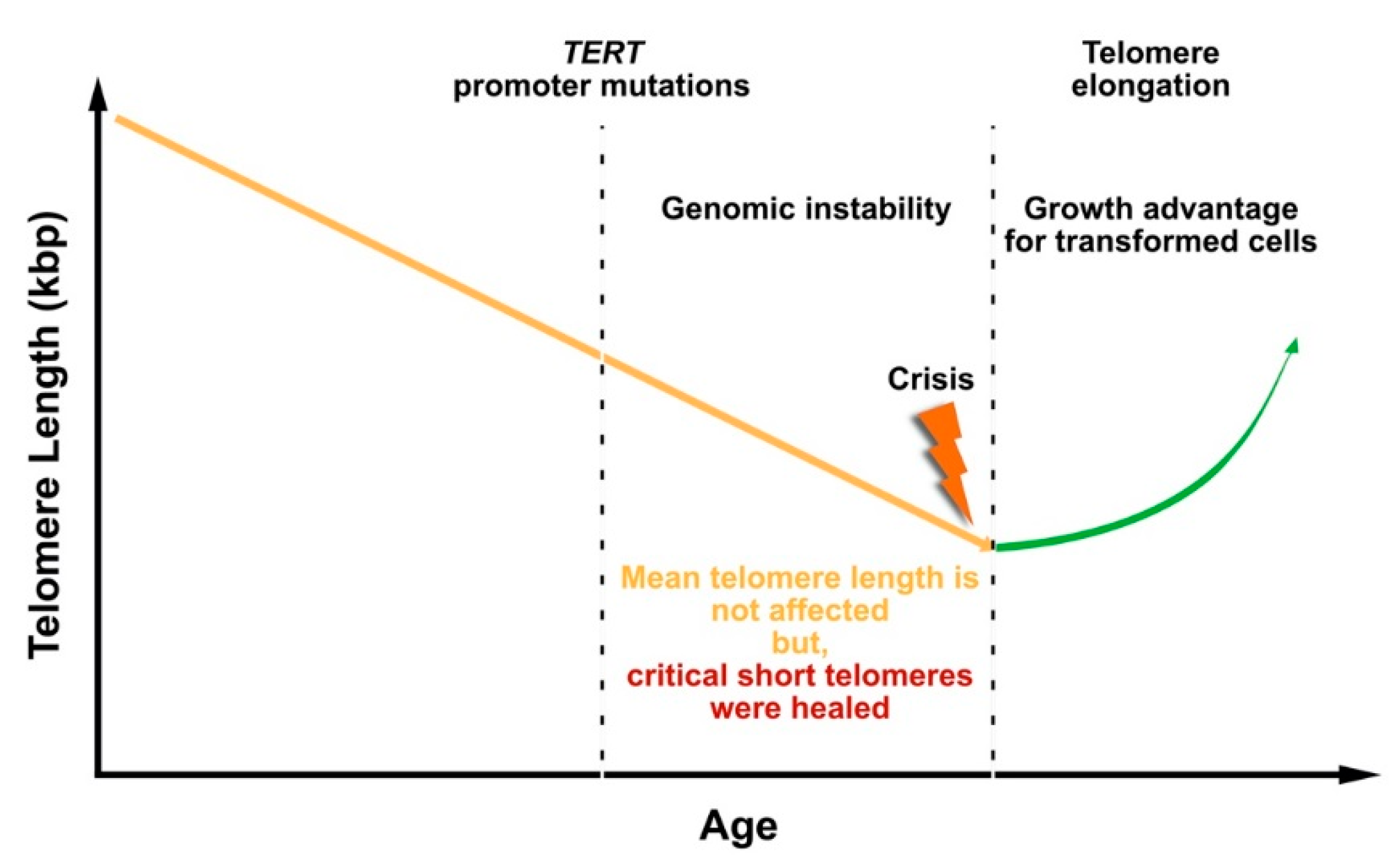
2. Telomere Shortening in Liver Cirrhosis and Hepatocellular Carcinoma
3. Mouse Models of Telomere Dysfunction in Hepatocarcinogenesis
4. Telomere Shortening in Cholangiocarcinoma
5. Loss of Function Mutations in Telomerase Components
6. Telomerase Reactivation during Hepatocarcinogenesis
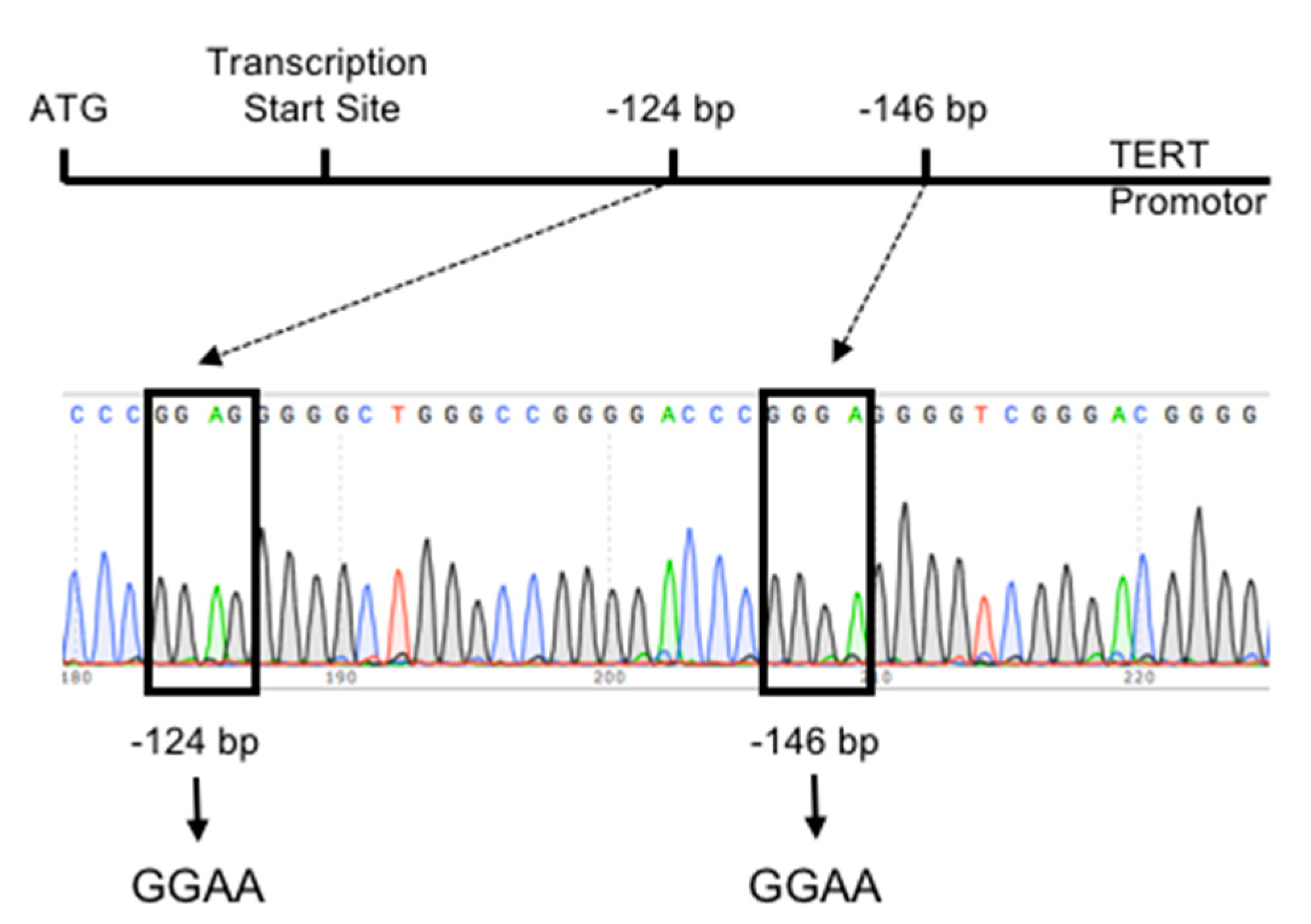
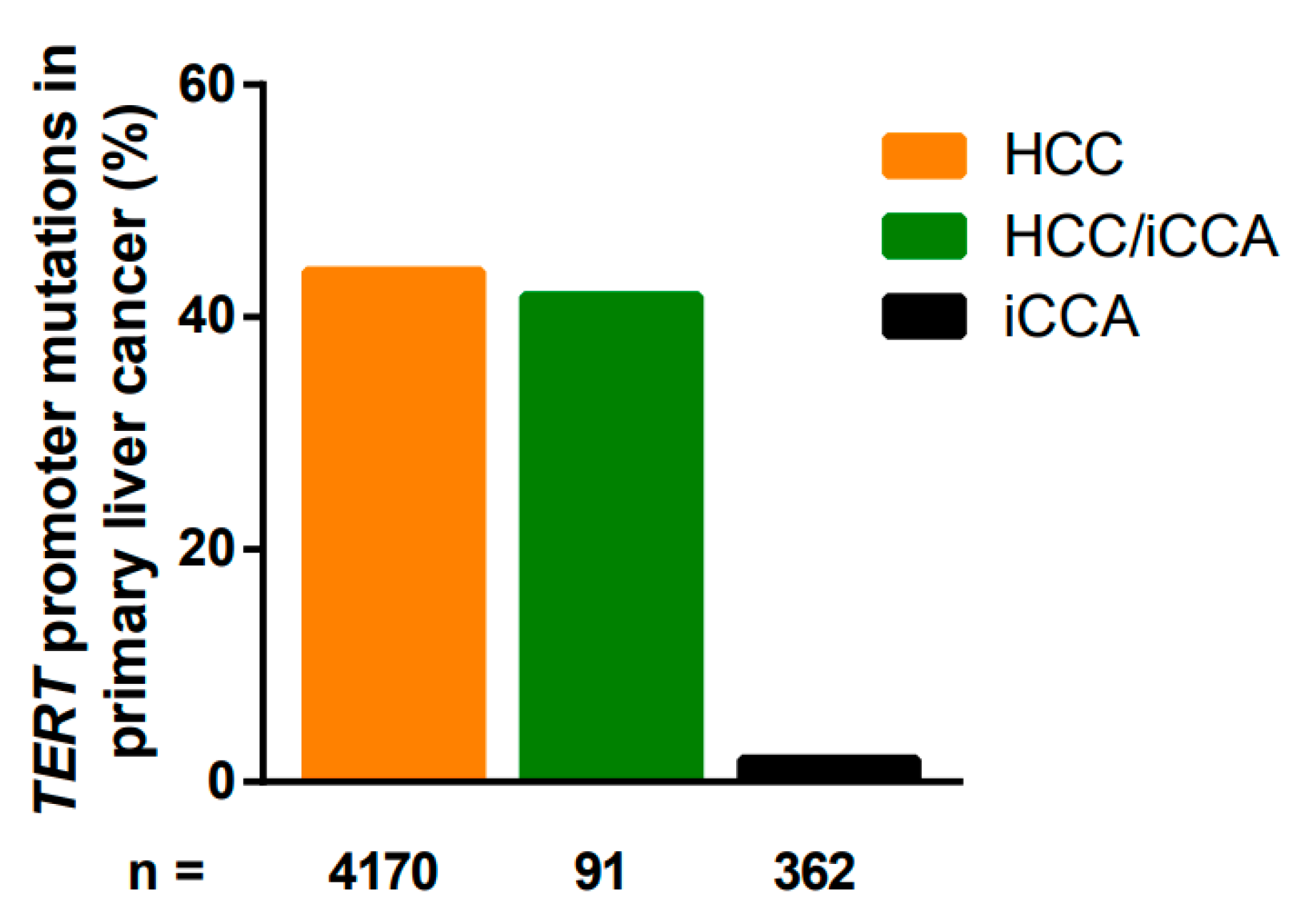
6.1. TERT Promoter Mutations
6.2. Amplification and Genomic Rearrangements of TERT
6.3. Altered Transcriptional Regulation of TERT Gene Expression
6.4. Epigenetic Mechanisms in the Regulation of TERT Gene Expression
7. Telomere and Telomerase-Based Cancer Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- London, W.T.; Petrick, J.L.; McGlynn, K.A. Liver Cancer. In Cancer Epidemiology and Prevention, 4th ed.; Thun, M.J., Linet, M.S., Cerhan, J.R., Haiman, C.A., Schottenfeld, D., Eds.; Oxford University Press: Cambridge, MA, USA, 2018; pp. 635–660. [Google Scholar]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16019. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular Carcinoma. J. Clin. Gastroenterol. 2002, 35, S72–S78. [Google Scholar] [CrossRef]
- Nault, J.-C. Pathogenesis of hepatocellular carcinoma according to aetiology. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 937–947. [Google Scholar] [CrossRef]
- Umemura, T.; Ichijo, T.; Yoshizawa, K.; Tanaka, E.; Kiyosawa, K. Epidemiology of hepatocellular carcinoma in Japan. J. Gastroenterol. 2009, 44, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2015, 22, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, H.; Patel, T. Molecular diagnosis of intrahepatic cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2014, 22, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC–aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yamaguchi, R.; Hasegawa, T.; Shimada, S.; Arihiro, K.; Hayashi, S.; Maejima, K.; Nakano, K.; Fujimoto, A.; Ono, A.; et al. Classification of primary liver cancer with immunosuppression mechanisms and correlation with genomic alterations. EBioMedicine 2020, 53, 102659. [Google Scholar] [CrossRef]
- Ahn, K.S.; O’Brien, D.; Kang, Y.N.; Mounajjed, T.; Kim, Y.H.; Kim, T.S.; Kocher, J.A.; Allotey, L.K.; Borad, M.J.; Roberts, L.R.; et al. Prognostic subclass of intrahepatic cholangiocarcinoma by integrative molecular-clinical analysis and potential targeted approach. Hepatol. Int. 2019, 13, 490–500. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, X.; Lu, X.; Luo, G.; Zeng, T.; Tang, J.; Jiang, F.; Li, L.; Cui, X.; Huang, W.; et al. Interferon-microRNA signalling drives liver precancerous lesion formation and hepatocarcinogenesis. Gut 2016, 65, 1186–1201. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Shay, J.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef]
- Kumar, M.; Lechel, A.; Günes, C. Telomerase: The Devil Inside. Genes 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; Depinho, R.A. Inhibition of Experimental Liver Cirrhosis in Mice by Telomerase Gene Delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Seki, S.; Kawakita, N.; Kuroki, T.; Monna, T. Telomere Shortening in Chronic Liver Diseases. Biochem. Biophys. Res. Commun. 1995, 211, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Urabe, Y.; Nouso, K.; Higashi, T.; Nakatsukasa, H.; Hino, N.; Ashida, K.; Kinugasa, N.; Yoshida, K.; Uematsu, S.; Tsuji, T. Telomere length in human liver diseases. Liver Int. 2008, 16, 293–297. [Google Scholar] [CrossRef]
- Paradis, V.; Youssef, N.; Dargere, D.; Bâ, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef]
- Mangnall, D.; Bird, N.C.; Majeed, A.W. The molecular physiology of liver regeneration following partial hepatectomy. Liver Int. 2003, 23, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Sirma, H.; Kumar, M.; Meena, J.K.; Witt, B.; Weise, J.M.; Lechel, A.; Ande, S.; Sakk, V.; Guguen-Guillouzo, C.; Zender, L.; et al. The Promoter of Human Telomerase Reverse Transcriptase Is Activated during Liver Regeneration and Hepatocyte Proliferation. Gastroenterology 2011, 141, 326–337.e3. [Google Scholar] [CrossRef]
- Kotoula, V.; Hytiroglou, P.; Pyrpasopoulou, A.; Saxena, R.; Thung, S.N.; Papadimitriou, C.S. Expression of human telomerase reverse transcriptase in regenerative and precancerous lesions of cirrhotic livers. Liver Int. 2002, 22, 57–69. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Chang, E.; Kashefi-Aazam, M.; Rogaev, E.I.; Piatyszek, M.A.; Shay, J.W.; Harley, C.B. Telomere Shortening Is Associated with Cell Division in Vitro and in Vivo. Exp. Cell Res. 1995, 220, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.; Gollahon, L.; Willingham, T.; Barbosa, M.; Shay, J. p53 levels in human mammary epithelial cells expressing wild-type and mutant human papillomavirus type 16 (HPV-16) E6 proteins. Int. J. Oncol. 1996, 8, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Brudno, J.; Mehta, P.; Kovacs, J.J.; Wu, C.; Zago, M.A.; Chanock, S.J.; Boyer, T.D.; Young, N.S. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011, 53, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, D.; Srivastava, U.; Thaler, M.; Kleinhans, K.N.; N’Kontchou, G.; Scheffold, A.; Bauer, K.; Kratzer, R.F.; Kloos, N.; Katz, S.-F.; et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011, 53, 1608–1617. [Google Scholar] [CrossRef] [Green Version]
- Tahara, H.; Nakanishi, T.; Kitamoto, M.; Nakashio, R.; Shay, J.W.; Tahara, E.; Kajiyama, G.; Ide, T. Telomerase activity in human liver tissues: Comparison between chronic liver disease and hepatocellular carcinomas. Cancer Res. 1995, 55, 2734–2736. [Google Scholar]
- Ide, T.; Tahara, H.; Nakashio, R.; Kitamoto, M.; Nakanishi, T.; Kajiyama, G. Telomerase in hepatocellular carcinogenesis. Hum. Cell 1996, 9, 283–286. [Google Scholar]
- Hytiroglou, P.; Kotoula, V.; Thung, S.N.; Tsokos, M.; Fiel, M.I.; Papadimitriou, C.S. Telomerase activity in precancerous hepatic nodules. Cancer 1998, 82, 1831–1838. [Google Scholar] [CrossRef]
- Youssef, N.; Paradis, V.; Ferlicot, S.; Bedossa, P. In situdetection of telomerase enzymatic activity in human hepatocellular carcinogenesis. J. Pathol. 2001, 194, 459–465. [Google Scholar] [CrossRef]
- Ozaki, S.; Harada, K.; Sanzen, T.; Watanabe, K.; Tsui, W.M.S.; Nakanuma, Y. In situ nucleic acid detection of human telomerase in intrahepatic cholangiocarcinoma and its preneoplastic lesion. Hepatology 1999, 30, 914–919. [Google Scholar] [CrossRef]
- Meena, J.; Rudolph, K.L.; Günes, C. Telomere Dysfunction, Chromosomal Instability and Cancer. Recent Results Cancer Res. 2015, 200, 61–79. [Google Scholar]
- Satyanarayana, A.; Manns, M.P.; Rudolph, K.L. Telomeres and telomerase: A dual role in hepatocarcinogenesis. Hepatology 2004, 40, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Park, Y.N.; Lechel, A.; Kim, H.; Nellessen, F.; Langkopf, B.H.E.; Wilkens, L.; Destro, A.; Fiamengo, B.; Manns, M.P.; et al. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology 2007, 45, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Caselitz, M.; Bleck, J.S.; Gebel, M.; Flemming, P.; Kubicka, S.; Manns, M.P.; Rudolph, K.L. Hepatocellular telomere shortening correlates with chromosomal instability and the development of human hepatoma. Hepatology 2004, 40, 80–86. [Google Scholar] [CrossRef]
- Plentz, R.R.; Schlegelberger, B.; Flemming, P.; Gebel, M.; Kreipe, H.; Manns, M.P.; Rudolph, K.L.; Wilkens, L. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology 2005, 42, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Denchi, E.L.; De Lange, T. Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; De Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Whole chromosome loss is promoted by telomere dysfunction in primary cells. Genes Chromosom. Cancer 2010, 49, 368–378. [Google Scholar] [CrossRef]
- Pampalona, J.; Frías, C.; Genesca, A.; Tusell, L. Progressive Telomere Dysfunction Causes Cytokinesis Failure and Leads to the Accumulation of Polyploid Cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef] [Green Version]
- Prowse, K.R.; Greider, C.W. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl. Acad. Sci. USA 1995, 92, 4818–4822. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Ritz, J.M.; Kühle, O.; Riethdorf, S.; Sipos, B.; Deppert, W.; Englert, C.; Gunes, C. A Novel Transgenic Mouse Model Reveals Humanlike Regulation of an 8-kbp Human TERT Gene Promoter Fragment in Normal and Tumor Tissues. Cancer Res. 2005, 65, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, I.; Chiang, Y.J.; Patterson, T.; Feigenbaum, L.; Leem, S.-H.; Michishita, E.; Larionov, V.; Hodes, R.J.; Barrett, J.C. Differential cis-regulation of human versus mouse TERT gene expression in vivo: Identification of a human-specific repressive element. Proc. Natl. Acad. Sci. USA 2005, 102, 18437–18442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolquist, K.A.; Ellisen, L.W.; Counter, C.M.; Meyerson, M.M.; Tan, L.K.; Weinberg, R.A.; Haber, D.A.; Gerald, W.L. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat. Genet. 1998, 19, 182–186. [Google Scholar] [CrossRef]
- Greenberg, R.A.; O’Hagan, R.C.; Deng, H.; Xiao, Q.; Hann, S.R.; Adams, R.R.; Lichtsteiner, S.; Chin, L.; Morin, G.B.; DePinho, R.A. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene 1999, 18, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Kipling, D.; Cooke, H.J. Hypervariable ultra-long telomeres in mice. Nature 1990, 347, 400–402. [Google Scholar] [CrossRef]
- Kipling, D. Telomere structure and telomerase expression during mouse development and tumorigenesis. Eur. J. Cancer 1997, 33, 792–800. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Wiemann, S.; Buer, J.; Lauber, J.; Dittmar, K.; Wüstefeld, T.; Blasco, M.A.; Manns, M.; Rudolph, K.L. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003, 22, 4003–4013. [Google Scholar] [CrossRef]
- Wiemann, S.U.; Satyanarayana, A.; Buer, J.; Kamino, K.; Manns, M.P.; Rudolph, K.L. Contrasting effects of telomere shortening on organ homeostasis, tumor suppression, and survival during chronic liver damage. Oncogene 2004, 24, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechel, A.; Holstege, H.; Begus, Y.; Schienke, A.; Kamino, K.; Lehmann, U.; Kubicka, S.; Schirmacher, P.; Jonkers, J.; Rudolph, K.L. Telomerase Deletion Limits Progression of p53-Mutant Hepatocellular Carcinoma With Short Telomeres in Chronic Liver Disease. Gastroenterology 2007, 132, 1465–1475. [Google Scholar] [CrossRef]
- Hemann, M.T. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000, 28, 4474–4478. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.A.; Gottlieb, G.J.; Greider, C.W.; Depinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003, 63, 5021–5027. [Google Scholar] [PubMed]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Begus-Nahrmann, Y.; Hartmann, D.; Kraus, J.; Eshraghi, P.; Scheffold, A.; Grieb, M.; Rasche, V.; Schirmacher, P.; Lee, H.-W.; Kestler, H.A.; et al. Transient telomere dysfunction induces chromosomal instability and promotes carcinogenesis. J. Clin. Investig. 2012, 122, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara-Romeo, I.; Martínez, P.; Blasco, M.A. Mice lacking RAP1 show early onset and higher rates of DEN-induced hepatocellular carcinomas in female mice. PLoS ONE 2018, 13, e0204909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Njei, B. Changing pattern of epidemiology in intrahepatic cholangiocarcinoma. Hepatology 2014, 60, 1107–1108. [Google Scholar] [CrossRef]
- Verma, S.; Tachtatzis, P.; Penrhyn-Lowe, S.; Scarpini, C.G.; Jurk, D.; Zglinicki, T.; Coleman, N.; Alexander, G.J.M. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology 2012, 56, 1510–1520. [Google Scholar] [CrossRef] [Green Version]
- Hansel, D.E.; Meeker, A.K.; Hicks, J.; De Marzo, A.M.; Lillemoe, K.D.; Schulick, R.; Hruban, R.H.; Maitra, A.; Argani, P. Telomere length variation in biliary tract metaplasia, dysplasia, and carcinoma. Mod. Pathol. 2006, 19, 772–779. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatològy 2008, 48, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.; Marrone, A.; Goldman, F.; Dearlove, A.; Bessler, M.; Mason, P.J.; Dokal, I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 2001, 413, 432–435. [Google Scholar] [CrossRef]
- Fogarty, P.F.; Yamaguchi, H.; Wiestner, A.; Baerlocher, G.M.; Sloand, E.; Zeng, W.; Read, E.J.; Lansdorp, P.M.; Young, N.S. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 2003, 362, 1628–1630. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Calado, R.T.; Ly, H.; Kajigaya, S.; Baerlocher, G.M.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. Mutations inTERT, the Gene for Telomerase Reverse Transcriptase, in Aplastic Anemia. N. Engl. J. Med. 2005, 352, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Regal, J.A.; Hills, M.; Yewdell, W.T.; Dalmazzo, L.F.; Zago, M.A.; Lansdorp, P.M.; Hogge, N.; Chanock, S.J.; Estey, E.H.; et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Kirwan, M.; Vulliamy, T.; Marrone, A.; Walne, A.J.; Beswick, R.; Hillmen, P.; Kelly, R.; Stewart, A.; Bowen, D.; Schönland, S.; et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum. Mutat. 2009, 30, 1567–1573. [Google Scholar] [CrossRef]
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017, 6, 1930–1940. [Google Scholar] [CrossRef]
- Barthel, F.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Zheng, C.; Lindvall, C.; Hou, M.; Ekedahl, J.; Lewensohn, R.; Yan, Z.; Yang, X.; Henriksson, M.; Blennow, E.; et al. Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res. 2000, 60, 6230–6235. [Google Scholar]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Kumar, M.; Witt, B.; Knippschild, U.; Koch, S.; Meena, J.K.; Heinlein, C.; Weise, J.M.; Krepulat, F.; Kuchenbauer, F.; Iben, S.; et al. CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int. J. Cancer 2013, 132, 2032–2043. [Google Scholar] [CrossRef]
- Günes, C.; Wezel, F.; Southgate, J.; Bolenz, C. Implications of TERT promoter mutations and telomerase activity in urothelial carcinogenesis. Nat. Rev. Urol. 2018, 15, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metab. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef] [Green Version]
- Rachakonda, P.S.; Hosen, I.; De Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro-Oncol. 2014, 17, 45–52. [Google Scholar] [CrossRef]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Chang, S.-H.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016, 7, 69267–69275. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [Green Version]
- Quaas, A.; Oldopp, T.; Tharun, L.; Klingenfeld, C.; Krech, T.; Sauter, G.; Grob, T.J. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014, 465, 673–677. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Chianchiano, P.; Pezhouh, M.K.; Kim, A.; Luchini, C.; Cameron, A.; Weiss, M.J.; He, J.; Voltaggio, L.; Oshima, K.; Anders, R.A.; et al. Distinction of intrahepatic metastasis from multicentric carcinogenesis in multifocal hepatocellular carcinoma using molecular alterations. Hum. Pathol. 2017, 72, 127–134. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2015, 51, 473–486. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.-J.; Jung, S.-J.; Lee, J.-H. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Kwa, W.T.; Effendi, K.; Yamazaki, K.; Kubota, N.; Hatano, M.; Ueno, A.; Masugi, Y.; Sakamoto, M. Telomerase reverse transcriptase (TERT) promoter mutation correlated with intratumoral heterogeneity in hepatocellular carcinoma. Pathol. Int. 2020. [Google Scholar] [CrossRef]
- Rudini, N.; Novello, C.; Destro, A.; Riboldi, E.; Donadon, M.; Viganò, L.; Morenghi, E.; Roncalli, M.; Di Tommaso, L. Phenotypic and molecular changes in nodule-in-nodule hepatocellular carcinoma with pathogenetic implications. Histopathology 2018, 73, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franzè, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [PubMed]
- Joseph, N.M.; Umetsu, S.E.; Shafizadeh, N.; Ferrell, L.; Kakar, S. Genomic profiling of well-differentiated hepatocellular neoplasms with diffuse glutamine synthetase staining reveals similar genetics across the adenoma to carcinoma spectrum. Mod. Pathol. 2019, 32, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Cheng, G.; Yu, J.; Zheng, S.; Sun, C.; Sun, Q.; Li, K.; Lin, Z.; Liu, T.; Li, P.; et al. The TERT promoter mutation incidence is modified by germline TERT rs2736098 and rs2736100 polymorphisms in hepatocellular carcinoma. Oncotarget 2017, 8, 23120–23129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilati, C.; Letouzé, E.; Nault, J.-C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic Profiling of Hepatocellular Adenomas Reveals Recurrent FRK-Activating Mutations and the Mechanisms of Malignant Transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhou, W.; Li, C.; Yang, Y.; Shang, Y.-K.; Chen, C.; Zhang, J.; Yao, R.; Wang, P.; Wen, W.; et al. Promoter mutations and cellular distribution of telomerase in non-clear cell and clear cell hepatocellular carcinoma. Oncotarget 2017, 8, 26288–26297. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.-F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jeng, Y.M.; Chang, C.N.; Lee, H.J.; Hsu, H.C.; Lai, P.L.; Yuan, R.-H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Ajiki, T.; Fukumoto, T.; Itoh, T.; Zen, Y. Keratin 19-expressing hepatocellular carcinoma and small-duct type intrahepatic cholangiocarcinoma show a similar postoperative clinical course but have distinct genetic features. Histopathology 2019, 75, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef] [Green Version]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Wu, S.C.; Chang, S.C.; Wu, H.Y.; Liao, P.J.; Chang, M.F. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 2008, 283, 29396–29404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.-C.; Zucman-Rossi, J. TERT promoter mutations in primary liver tumors. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 9–14. [Google Scholar] [CrossRef]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.-I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Joseph, N.M.; Tsokos, C.G.; Umetsu, S.E.; Shain, A.H.; Kelley, R.K.; Onodera, C.; Bowman, S.; Talevich, E.; Ferrell, L.D.; Kakar, S.; et al. Genomic profiling of combined hepatocellular-cholangiocarcinoma reveals similar genetics to hepatocellular carcinoma. J. Pathol. 2019, 248, 164–178. [Google Scholar] [CrossRef]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Mutational landscape of combined hepatocellular carcinoma and cholangiocarcinoma, and its clinicopathological significance. Histopathology 2016, 70, 423–434. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Kratz, J.; Brat, D.J.; Robitaille, Y.; Moghrabi, A.; Perlman, E.J.; Dang, C.V.; Burger, P.C.; Eberhart, C.G. hTERT Gene Amplification and Increased mRNA Expression in Central Nervous System Embryonal Tumors. Am. J. Pathol. 2003, 162, 1763–1769. [Google Scholar] [CrossRef] [Green Version]
- Takuma, Y.; Nouso, K.; Kobayashi, Y.; Nakamura, S.; Tanaka, H.; Matsumoto, E.; Fujikawa, T.; Suzuki, M.; Hanafusa, T.; Shiratori, Y. Telomerase reverse transcriptase gene amplification in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 1300–1304. [Google Scholar] [CrossRef]
- Palmqvist, R.; Zhang, A.; Xu, D.; Golovleva, I.; Norrback, K.-F.; Gruber, A.; Öberg, Å.; Stenling, R.; Roos, G. hTERT gene copy number is not associated with hTERT RNA expression or telomerase activity in colorectal cancer. Int. J. Cancer 2005, 116, 395–400. [Google Scholar] [CrossRef]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; Van Sluis, P.; Volckmann, R.; Van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Meric-Bernstam, F.; Medeiros, L.J.; Weinstein, J.N.; et al. Landscape of DNA Virus Associations across Human Malignant Cancers: Analysis of 3,775 Cases Using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Logan, G.J.; Dane, A.P.; Hallwirth, C.V.; Smyth, C.M.; Wilkie, E.E.; Amaya, A.K.; Zhu, E.; Khandekar, N.; Ginn, S.L.; Liao, S.H.Y.; et al. Identification of liver-specific enhancer–promoter activity in the 3′ untranslated region of the wild-type AAV2 genome. Nat. Genet. 2017, 49, 1267–1273. [Google Scholar] [CrossRef]
- Choi, S.H.; Cho, K.J.; Yun, S.H.; Jin, B.; Lee, H.Y.; Ro, S.W.; Kim, D.Y.; Ahn, S.H.; Han, K.-H.; Park, J.Y. HKR3 regulates cell cycle through the inhibition of hTERT in hepatocellular carcinoma cell lines. J. Cancer 2020, 11, 2442–2452. [Google Scholar] [CrossRef]
- Liu, T.; Li, W.; Lu, W.; Chen, M.; Luo, M.; Zhang, C.; Li, Y.; Qin, G.; Shi, D.; Xiao, B.; et al. RBFOX3 Promotes Tumor Growth and Progression via hTERT Signaling and Predicts a Poor Prognosis in Hepatocellular Carcinoma. Theranostics 2017, 7, 3138–3154. [Google Scholar] [CrossRef]
- Ko, E.; Seo, H.-W.; Jung, E.S.; Kim, B.-H.; Jung, G. The TERT promoter SNP rs2853669 decreases E2F1 transcription factor binding and increases mortality and recurrence risks in liver cancer. Oncotarget 2015, 7, 684–699. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, F.; Li, Y.; Hao, J.; Tang, Z.; Tian, C.; Yang, Q.; Zhu, T.; Diao, C.; Zhang, C.; et al. BPTF promotes hepatocellular carcinoma growth by modulating hTERT signaling and cancer stem cell traits. Redox Boil. 2019, 20, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Günes, C.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the hTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res. 2000, 60, 2116–2121. [Google Scholar] [PubMed]
- Wang, J.; Xie, L.Y.; Allan, S.; Beach, D.; Hannon, G.J. Myc activates telomerase. Genes Dev. 1998, 12, 1769–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.-J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [Green Version]
- Abou-Elella, A.; Gramlich, T.; Fritsch, C.; Gansler, T. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod. Pathol. 1996, 9, 95–98. [Google Scholar]
- Takahashi, Y.; Kawate, S.; Watanabe, M.; Fukushima, J.-I.; Mori, S.; Fukusato, T. Amplification of c-myc and cyclin D1 genes in primary and metastatic carcinomas of the liver. Pathol. Int. 2007, 57, 437–442. [Google Scholar] [CrossRef]
- Amisaki, M.; Tsuchiya, H.; Sakabe, T.; Fujiwara, Y.; Shiota, G. Identification of genes involved in the regulation of TERT in hepatocellular carcinoma. Cancer Sci. 2019, 110, 550–560. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Sowa, Y.; Wang, Z.; Yatabe, N.; Maida, Y.; Tanaka, M.; Inoue, M. Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res. 2001, 29, 3006–3011. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.; Wang, X.; Popov, N.; Zhang, A.; Zhao, X.; Zhou, R.; Zetterberg, A.; Björkholm, M.; Henriksson, M.; Gruber, A.; et al. The Histone Deacetylase Inhibitor Trichostatin A Derepresses the Telomerase Reverse Transcriptase (hTERT) Gene in Human Cells. Exp. Cell Res. 2002, 274, 25–34. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Yu, J.; Yuan, X.; Sjöholm, L.; Liu, T.; Kong, F.; Ekström, T.J.; Björkholm, M.; Xu, D. Telomerase reverse transcriptase regulates DNMT3B expression/aberrant DNA methylation phenotype and AKT activation in hepatocellular carcinoma. Cancer Lett. 2018, 434, 33–41. [Google Scholar] [CrossRef]
- Devereux, T.R.; Horikawa, I.; Anna, C.H.; Annab, L.A.; Afshari, C.A.; Barrett, J.C. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999, 59, 6087–6090. [Google Scholar]
- Dessain, S.K.; Yu, H.; Reddel, R.R.; Beijersbergen, R.L.; Weinberg, R.A. Methylation of the human telomerase gene CpG island. Cancer Res. 2000, 60, 537–541. [Google Scholar]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef]
- Zhang, H.; Weng, X.; Ye, J.; He, L.; Zhou, D.; Liu, Y. Promoter hypermethylation of TERT is associated with hepatocellular carcinoma in the Han Chinese population. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Satra, M.; Drakaki, A.; Poultsides, G.A.; Tsezou, A. Epigenetic regulation of hTERT promoter in hepatocellular carcinomas. Int. J. Oncol. 2009, 34, 391–399. [Google Scholar]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 1801. [Google Scholar] [CrossRef]
- Esopi, D.; Graham, M.K.; Brosnan-Cashman, J.A.; Meyers, J.; Vaghasia, A.; Gupta, A.; Kumar, B.; Haffner, M.C.; Heaphy, C.M.; De Marzo, A.M.; et al. Pervasive promoter hypermethylation of silenced TERT alleles in human cancers. Cell. Oncol. 2020. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Günes, C.; Rudolph, K.L. Telomere dysfunction putss the brakes on oncogene-induced cancers. EMBO J. 2012, 31, 2833–2904. [Google Scholar] [CrossRef]
- Günes, C.; Rudolph, K.L. The Role of Telomeres in Stem Cells and Cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| HCC * | 61 | 44.2% (27/61) | HCV | Killela et al., 2013 [96] | |
| 62.5% (10/16) | 0 (0/16) | ||||
| HBV | |||||
| 26.6% (4/15) | 0 (0/15) | ||||
| ETOH | |||||
| 100% (2/2) | 0 | ||||
| cyptogenic liver disease | |||||
| 100% (1/1) | 0 | ||||
| unknown | |||||
| 50% (8/16) | 0 (0/16) | ||||
| HCC | 70 # | 71% (50/70) | HCV | Chianchiano et al., 2018 [97] | |
| 87.5% (35/40) | 2.5% (1/40) | ||||
| HBV | |||||
| 0 (0/7) | 0 (0/7) | ||||
| ETOH | |||||
| 16.6% (1/6) | 0 (0/6) | ||||
| HCV/HBV | |||||
| 100% (2/2) | 0 | ||||
| unknown | |||||
| 73.3% (11/15) | 0 (0/15) | ||||
| HCC | 457 | 54.2% (248/457) | HCV | Totoki et al., 2014 [98] | |
| 62.2% (117/188) | 1.6% (3/188) | ||||
| HBV | |||||
| 28.7% (31/108) | 3.7% (4/108) | ||||
| HCV/HBV | |||||
| 66.6% (8/12) | 0 (0/12) | ||||
| NBNC † | |||||
| 53.6% (80/149) | 3.3% (5/149) | ||||
| HCC | 104 | 65% ‡ (68/104) | HCV | Kawai-Kitahata et al., 2016 [99] | |
| 80% (40/50) | |||||
| HBV | |||||
| 32% (9/28) | |||||
| ETOH | |||||
| 83% (10/12) | |||||
| unknown | |||||
| 64% (9/14) | |||||
| HCC | 160 | 28.8% § (46/160) | HCV | Lee et al., 2017 [100] | |
| 60% (3/5) | |||||
| HBV | |||||
| 32.7% (19/58) | |||||
| ETOH | |||||
| 28.5% (6/21) | |||||
| others | |||||
| 23.6% (18/76) | |||||
| HCC | 105 | 39% (41/105) | HCV | Lee et al., 2016 [93] | |
| 83.3% (5/6) | |||||
| HBV | |||||
| 29.4% (23/78) | |||||
| ETOH | |||||
| 37.5% (3/8) | |||||
| unknown | |||||
| 76.9% (10/13) | |||||
| HCC | 44 | 34% (15/44) | HBV | Cevik et al., 2015 [101] | |
| 13% (3/23) | 13% (3/23) | ||||
| unknown | |||||
| 33.3% (7/21) | 9.5% (2/21) | ||||
| HCC | 97 | 54.6% (53/97) | HCV | Kwa et al., 2020 [102] | |
| 71% (22/31) | |||||
| HBV | |||||
| 36.4% (8/22) | |||||
| NBNC | |||||
| 52.3% (23/44) | |||||
| HCC | 10 | 50% (5/10) | HCV | Rudini et al., 2018 [103] | |
| 42.9% (3/7) | 14.3% (1/7) | ||||
| ETOH | |||||
| 0 (0/2) | 0 (0/2) | ||||
| unknown | |||||
| 100% (1/1) | 0 (0/1) | ||||
| HCC | 67 | 43.3% (29/67) | ESC-NA | Lombardo et al., 2020 [104] | |
| HCC | 14 | 21.4% (3/14) | ESC-NA | Jospeh et al., 2019 [105] | |
| HCC | 127 | 50.4% (64/127) | HCV | Pezzuto et al., 2016 [106] | |
| 53.6% (59/110) | |||||
| HBV | |||||
| 41.7% (5/12) | |||||
| NBNC | |||||
| 0 (0/5) | |||||
| HCC | 11 | 81.9% (9/11) | NAFLD | Kim et al., 2016 [107] | |
| 100% (9/9) | 0 (0/9) | ||||
| HCC | 190 | 30% (57/190) | HBV | Yuan et al., 2017 [108] | |
| 32.7% (50/153) | |||||
| unknown | |||||
| 18.9% (7/37) | |||||
| HCC | 375 | 20.3% (76/375) | classical HCC | Pilati et al., 2014 [109] | |
| 54% (68/125) | |||||
| HCC derived from adenomas | |||||
| 56% (5/9) | |||||
| borderline lesions HCA/HCC | |||||
| 17% (3/18) | |||||
| classical adenomas | |||||
| 0 (0/223) | |||||
| HCC | 196 | 44.4% (87/196) | HCV | The Cancer Genome Atlas Research Network 2017 [110] | |
| 61.3% (19/31) | 3.2% (1/31) | ||||
| HBV | |||||
| 22.5% (9/40) | 2.5% (1/40) | ||||
| HCV/HBV | |||||
| 50% (2/4) | 0 (0/4) | ||||
| NBNC | |||||
| 40.5% (49/121) | 5% (6/121) | ||||
| HCC | 88 | 29.6% (26/88) | low-grade dysplastic nodules | Nault et al., 2014 [117] | |
| 6.3% (2/32) | |||||
| high-grade dysplastic nodules | |||||
| 18.8% (3/16) | |||||
| early HCC | |||||
| 60.9% (14/23) | |||||
| progressed HCC | |||||
| 41.2% (7/17) | |||||
| HCC | 276 | 31% (85/276) | HBV | Yang et al., 2016 [111] | |
| 98.8% (84/85) | 1.2% (1/85) | ||||
| non-clear cell HCC | 259 | 33.2% (86/259) | 94.2% (81/86) | 5.8% (5/86) | Huang et al., 2017 [112] |
| clear cell HCC | 57 | 26.3% (15/57) | 100% (15/15) | 0 (0/15) | |
| HCC | 322 | 64.5% (208/322) | combined etiology | Calderaro et al., 2017 [113] | |
| 64.5% (208/322) | |||||
| HCC | 195 | 29.2% (57/195) | 94.7% (54/57) | 5.3% (3/57) | Chen et al., 2014 [114] |
| HCC | 235 | 60.4% (142/235) | combined etiology | Schulze et al., 2015 [115] | |
| 60.4% (142/235) | |||||
| HCC | 35 ** | 31.4% (11/35) | 81.8% (9/11) | 18.2% (2/11) | Huang et al., 2015 [92] |
| HCC | 78 ** | 47% (37/78) | 100% (37/37) | 0 (0/37) | Quaas et al., 2014 [95] |
| HCC | 305 ** | 58.6% (179/305) | 93.8% (168/179) | 6.1% (11/179) | Nault et al., 2013 [94] |
| HCC | 162 ** | 45% (73/162) | NA | NA | Barthel et al., 2017 [82] |
| HCC (K19−) | 44 *** | 59% (26/44) | 100% (26/26) | 0 | Akita et al., 2019 [116] |
| HCC (K19+) | 26 *** | 31% (8/26) | 100% (8/8) | 0 | |
| Total | 4170 | 43.9% (1831/4170) | |||
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| iCCA | 145 | 0.70% (1/145) | HCV | Nakamura et al., 2015 [125] | |
| 0 (0/10) | 0 (0/10) | ||||
| HBV | |||||
| 0 (0/7) | 0 (0/7) | ||||
| NBNC † | |||||
| 0.8% (1/122) | 0 (0/122) | ||||
| unknown | |||||
| 0 (0/6) | 0 (0/6) | ||||
| iCCA | 78 | 5.12% (4/78) | HCV | Fujimoto et al., 2015 [124] | |
| 22.2% (2/9) | |||||
| HBV | |||||
| 9% (1/11) | |||||
| NBNC † | |||||
| 1.7% (1/58) | |||||
| iCCA | 10 | 10% (1/10) | HCV | Joseph et al., 2019 [126] | |
| 0 (0/5) | |||||
| HBV | |||||
| 0 (0/2) | |||||
| NASH | |||||
| 50% (1/2) | |||||
| PBC | |||||
| 0 (0/1) | |||||
| CC | 4 | 25% (1/4) | HCV | Pezzuto et al., 2016 [106] | |
| 25% (1/4) | |||||
| iCCA | 9 ** | 0 (0/9) | 0 (0/9) | 0 (0/9) | Huang et al., 2015 [92] |
| iCCA | 52 ** | 0 (0/52) | 0 (0/52) | 0 (0/52) | Quaas et al., 2014 [95] |
| iCCA | 28 ** | 0 (0/28) | 0 (0/28) | 0 (0/28) | Killela et al., 2013 [96] |
| S-iCCA | 36 *** | 0 (0/36) | 0 (0/36) | 0 (0/36) | Akita et al., 2019 [116] |
| Total | 362 | 1.9% (7/362) | |||
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| cHCC/CC | 15 | 53.3% (8/15) | HCV | Fujimoto et al., 2015 [124] | |
| 83.3% (5/6) | |||||
| HBV | |||||
| 0 (0/3) | |||||
| NBNC * | |||||
| 50% (3/6) | |||||
| combined HCC-CC | 20 | 70% (14/20) | HCV | Joseph et al., 2019 [126] | |
| 81.8% (9/11) | |||||
| HBV | |||||
| 0 (0/1) | |||||
| HCV/HBV | |||||
| 100% (1/1) | |||||
| ASH | |||||
| 100% (1/1) | |||||
| NASH | |||||
| 100% (1/1) | |||||
| ASH/NASH | |||||
| 0% (0/1) | |||||
| PSC | |||||
| 100% (1/1) | |||||
| unknown | |||||
| 33.3% (1/3) | |||||
| cHC-CC | 53 | 30.2% (16/53) | HCV | Sasaki et al., 2017 [127] | |
| 31.8% (7/22) | |||||
| HBV | |||||
| 44.5% (4/9) | |||||
| ETOH | |||||
| 40% (2/5) | |||||
| NAFLD | |||||
| 0 (0/8) | |||||
| unknown | |||||
| 33.4% (3/9) | |||||
| HCC-CC | 3 | 0 (0/3) | HCV | Pezzuto et al., 2016 [106] | |
| 0 (0/2) | |||||
| HBV | |||||
| 0 (0/1) | |||||
| Total | 91 | 41.8% (38/91) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
in der Stroth, L.; Tharehalli, U.; Günes, C.; Lechel, A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers 2020, 12, 2048. https://doi.org/10.3390/cancers12082048
in der Stroth L, Tharehalli U, Günes C, Lechel A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers. 2020; 12(8):2048. https://doi.org/10.3390/cancers12082048
Chicago/Turabian Stylein der Stroth, Lena, Umesh Tharehalli, Cagatay Günes, and André Lechel. 2020. "Telomeres and Telomerase in the Development of Liver Cancer" Cancers 12, no. 8: 2048. https://doi.org/10.3390/cancers12082048
APA Stylein der Stroth, L., Tharehalli, U., Günes, C., & Lechel, A. (2020). Telomeres and Telomerase in the Development of Liver Cancer. Cancers, 12(8), 2048. https://doi.org/10.3390/cancers12082048