Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Clinical Correlates of Spliceosome Mutations in MPN
3. Consequences of Splicing Anomalies in Myeloid Malignancies
3.1. Molecular Mechanisms of Spliceosome Mutations
3.2. Splicing Anomalies in Myeloid Malignancies, Independent of Spliceosome Mutations
4. Mechanisms Driving Splicing Anomalies in the Evolution of MPN
5. Therapeutic Targeting of Splicing Anomalies
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ren, R. Mechanisms of BCR–ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Jabbour, E.J.; Kantarjian, H.M. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.A.; Carroll, M. BCR–ABL: A multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia 2010, 24, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinfeld, J.; Nangalia, J.; Green, A.R. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 2016, 102, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- McWhirter, J.R.; Galasso, D.L.; Wang, J.Y. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Boil. 1993, 13, 7587–7595. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fouroucls, N.; Swanton, S.; Vassilou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.; Teo, S.-S.; Tiedt, R.; Passweg, J.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, 270. [Google Scholar] [CrossRef] [Green Version]
- Chaligné, R.; James, C.; Tonetti, C.; Besancenot, R.; Le Couédic, J.P.; Fava, F.; Mazurier, F.; Godin, I.; Maloum, K.; Larbret, F.; et al. Evidence for MPL W515L/K mutations in hematopoietic stem cells in primitive myelofibrosis. Blood 2007, 110, 3735–3743. [Google Scholar] [CrossRef]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Them, N.C.C.; Berg, T.; Vladimer, G.I.; Bagienski, K.; Milanesi, C.; Casetti, I.C.; Sant’Antonio, E.; Ferretti, V.V.; Schischlik, F.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Balligand, T.; Achouri, Y.; Pecquet, C.; Chachoua, I.; Nivarthi, H.; Marty, C.; Vainchenker, W.; Plo, I.; Kralovics, R.; Constantinescu, S.N. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia 2016, 30, 1775–1778. [Google Scholar] [CrossRef] [Green Version]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, C.; Pecquet, C.; Nivarthi, H.; El-Khoury, M.; Tulliez, M.; Villeval, J.-L.; Raslova, H.; Kralovics, R.; Constantinescu, S.N.; Plo, I.; et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016, 127, 1317–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivarthi, H.; Chen, D.; Cleary, C.; Kubesova, B.; Jäger, R.; Bogner, E.; Marty, C.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N.; et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 2016, 30, 1759–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Green, A.R.; Beer, P. Somatic Mutations of IDH1 and IDH2 in the Leukemic Transformation of Myeloproliferative Neoplasms. N. Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, B.; Akinyi, M.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Boil. 2018, 79, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Splicing of a rare class of introns by the U12-dependent spliceosome. Boil. Chem. 2005, 386, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/proteomes/UP000005640 (accessed on 29 June 2020).
- Harrow, J.; Frankish, A.; González, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Pawlicka, K.; Kalathiya, U.; Alfaro, J.A. Nonsense-Mediated mRNA Decay: Pathologies and the Potential for Novel Therapeutics. Cancers 2020, 12, 765. [Google Scholar] [CrossRef] [Green Version]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. Bioessays 2017, 39, 1600157. [Google Scholar] [CrossRef]
- Cáceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Holm, F.; Hellqvist, E.; Mason, C.N.; Ali, S.A.; Delos-Santos, N.; Barret, C.L.; Chun, H.-J.; Minden, M.D.; Moore, R.A.; Mara, M.A.; et al. Reversion to an embryonic alternative splicing program enhances leukemia stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2015, 112, 15444–15449. [Google Scholar] [CrossRef] [Green Version]
- Luco, R.F.; Alló, M.; Schor, I.E.; Kornblihtt, A.R.; Misteli, T. Epigenetics in Alternative Pre-mRNA Splicing. Cell 2011, 144, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Adamia, S.; Haibe-Kains, B.; Pilarski, P.M.; Bar-Natan, M.; Pevzner, S.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; Fox, E.A.; Burke, J.; et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin. Cancer Res. 2013, 20, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anande, G.; Deshpande, N.P.; Mareschal, S.; Batcha, A.M.N.; Hampton, H.R.; Herold, T.; Lehmann, S.; Wilkins, M.R.; Wong, J.W.H.; Unnikrishnan, A.; et al. RNA Splicing Alterations Induce a Cellular Stress Response Associated with Poor Prognosis in Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 3597–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer 2019, 59, 30–39. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Wassie, E.A.; Knudson, R.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A. U2AF1 mutations in primary myelofibrosis are strongly associated with anemia and thrombocytopenia despite clustering with JAK2V617F and normal karyotype. Leukemia 2013, 28, 431–433. [Google Scholar] [CrossRef]
- Venton, G.; Courtier, F.; Charbonnier, A.; D’Incan, E.; Saillard, C.; Mohty, B.; Mozziconacci, M.-J.; Birnbaum, D.; Murati, A.; Vey, N.; et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am. J. Hematol. 2017, 93, 330–338. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Pacilli, A.; Rotunno, G.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017, 129, 3227–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Boiocchi, L.; Hasserjian, R.P.; Pozdnyakova, O.A.; Wong, W.J.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, R.; Iafrate, A.J.; Hobbs, G.S.; Nardi, V. Clinicopathological and molecular features of SF3B1-mutated myeloproliferative neoplasms. Hum. Pathol. 2019, 86, 1–11. [Google Scholar] [CrossRef]
- Barraco, D.; Elala, Y.C.; Lasho, T.L.; Begna, K.L.; Gangat, N.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardnani, A.; Tefferi, A. Molecular correlates of anemia in primary myelofibrosis: A significant and independent association with U2AF1 mutations. Blood Cancer J. 2016, 6, e415. [Google Scholar] [CrossRef]
- Byun, J.M.; Song, S.; Koh, Y.; Yoon, S.-S.; Kim, D. The Temporal Sequence and the Differences in Somatic Mutation Acquisition Determines Clinical Behaviors of JAK2-Positive Myeloproliferative Neoplasms. Anticancer Res. 2019, 39, 6273–6282. [Google Scholar] [CrossRef]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematology 2016, 102, e11–e14. [Google Scholar] [CrossRef]
- Delic, S.; Rose, M.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef]
- Li, B.; Gale, R.P.; Xu, Z.; Qin, T.; Song, Z.; Zhang, P.; Bai, J.; Zhang, L.; Zhang, Y.; Liu, J.; et al. Non-driver mutations in myeloproliferative neoplasm-associated myelofibrosis. J. Hematol. Oncol. 2017, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2011, 26, 1135–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, D.L.; Chauveau, A.; Boyer, F.; Buors, C.; Samaison, L.; Cottin, L.; Seegers, V.; Férec, C.; Le Maréchal, C.; Gueguen, P.; et al. Sequential analysis of 18 genes in polycythemia vera and essential thrombocythemia reveals an association between mutational status and clinical outcome. Genes Chromosomes Cancer 2017, 56, 354–362. [Google Scholar] [CrossRef]
- Brecqueville, M.; Rey, J.; Bertucci, F.; Coppin, E.; Finetti, P.; Garbuccia, N.; Cervera, N.; Gelsi-Boyer, V.; Arnoulet, C.; Gisserot, O.; et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer 2012, 51, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, M.M.; Lasho, T.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.; Gangat, N.; Tefferi, A. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am. J. Hematol. 2016, 91, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeromin, S.; Haferlach, T.; Grossmann, V.; Alpermann, T.; Kowarsch, A.; Haferlach, C.; Kern, W.; Schnittger, S. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica 2012, 98, e15–e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federmann, B.; Abele, M.; Cuesta, D.S.R.; Vogel, W.; Boiocchi, L.; Kanz, L.; Quintanilla-Martinez, L.; Orazi, A.; Bonzheim, I.; Fend, F. The detection of SRSF2 mutations in routinely processed bone marrow biopsies is useful in the diagnosis of chronic myelomonocytic leukemia. Hum. Pathol. 2014, 45, 2471–2479. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Jacqueline, B.; Bowen, D.T.; Campbell, B.L.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.; Hellström-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Quesada, V.; Conde, L.; Villamor, N.; Ordoñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhou, B.; Thol, F.; Zhou, Y.; Chen, L.; Shao, C.; DeBoever, C.; Hou, J.; Li, H.; Chaturvedi, A.; et al. Distinct splicing signatures affect converged pathways in myelodysplastic syndrome patients carrying mutations in different splicing regulators. RNA 2016, 22, 1535–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, J.M.; Sanford, J. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2014, 6, 93–110. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossmann, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S.; et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012, 120, 3080–3088. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [Green Version]
- Pangallo, J.; Kiladjian, J.-J.; Cassinat, B.; Renneville, A.; Taylor, J.; Polaski, J.T.; North, K.D.; Abdel-Wahab, O.; Bradley, R.K. Rare and private spliceosomal gene mutations drive partial, complete, and dual phenocopies of hotspot alterations. Blood 2020, 135, 1032–1043. [Google Scholar] [CrossRef]
- Liang, Y.; Tebaldi, T.; Rejeski, K.; Joshi, P.; Stefani, G.; Taylor, A.; Song, Y.; Vasic, R.; Maziarz, J.; Balasubramanian, K.; et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 2018, 32, 2659–2671. [Google Scholar] [CrossRef]
- Smeets, M.F.; Tan, S.Y.; Xu, J.J.; Anande, G.; Unnikrishnan, A.; Chalk, A.M.; Taylor, S.R.; Pimanda, J.E.; Wall, M.; Purton, L.E.; et al. Srsf2 P95H initiates myeloid bias and myelodysplastic/myeloproliferative syndrome from hemopoietic stem cells. Blood 2018, 132, 608–621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadán, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef] [PubMed]
- Kon, A.; Yamazaki, S.; Nannya, Y.; Kataoka, K.; Ota, Y.; Nakagawa, M.; Yoshida, K.; Shiozawa, Y.; Morita, M.; Yoshizato, T.; et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2018, 131, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Lee, S.C. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer 2019, 58, 889–902. [Google Scholar] [CrossRef]
- Przychodzen, B.; Jerez, A.; Guinta, K.; Sekeres, M.A.; Padgett, R.; Maciejewski, J.P.; Makishima, H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 2013, 122, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.J.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2014, 25, 14–26. [Google Scholar] [CrossRef]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nature 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Fei, D.L.; Zhen, T.; Durham, B.H.; Ferrarone, J.; Zhang, T.; Garrett, L.; Yoshimi, A.; Abdel-Wahab, O.; Bradley, R.K.; Liu, P.P.; et al. Impaired hematopoiesis and leukemia development in mice with a conditional knock-in allele of a mutant splicing factor gene U2af1. Proc. Natl. Acad. Sci. USA 2018, 115, E10437–E10446. [Google Scholar] [CrossRef] [Green Version]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Cola, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wasef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neuman, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Feenstra, J.D.M.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, D.; Chew, G.-L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.-W.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nature 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Mupo, A.; Seiler, M.; Sathiaseelan, V.; Pance, A.; Yang, Y.; Agrawal, A.A.; Iorio, F.; Bautista, R.; Pacharne, S.; Tzelepis, K.; et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia 2016, 31, 720–727. [Google Scholar] [CrossRef]
- Adamia, S.; Bar-Natan, M.; Haibe-Kains, B.; Pilarski, P.M.; Bach, C.; Pevzner, S.; Calimeri, T.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; et al. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): New potential targets in AML. Blood 2014, 123, 2816–2825. [Google Scholar] [CrossRef] [Green Version]
- Crews, L.; Balaian, L.; Santos, N.D.; Leu, H.S.; Court, A.C.; Lazzari, E.; Sadarangani, A.; Zipeto, M.A.; La Clair, J.J.; Villa, R.; et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell 2016, 19, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsson, A.E.; Geron, I.; Gotlib, J.; T Dao, K.-H.; Barroga, C.F.; Newton, I.G.; Giles, J.F.; Durocher, J.; Creusot, R.S.; Karimi, M.; et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc. Natl. Acad. Sci. USA 2009, 106, 3925–3929. [Google Scholar] [CrossRef] [Green Version]
- Vassen, L.; Khandanpour, C.; Ebeling, P.; Van Der Reijden, B.A.; Jansen, J.H.; Mahlmann, S.; Dührsen, U.; Möröy, T. Growth factor independent 1b (Gfi1b) and a new splice variant of Gfi1b are highly expressed in patients with acute and chronic leukemia. Int. J. Hematol. 2009, 89, 422–430. [Google Scholar] [CrossRef]
- Goff, D.J.; Recart, A.C.; Sadarangani, A.; Chun, H.-J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, Y.; Li, S.-Q.; Fang, L.; Wang, X.-Z.; Li, J.; Zhang, H.-B.; Huang, B.; Xu, Y.-M.; Yang, W.-M.; et al. Correction of Bcl -x splicing improves responses to imatinib in chronic myeloid leukaemia cells and mouse models. Br. J. Haematol. 2020, 189, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Salesse, S.; Dylla, S.J.; Verfaillie, C.M. p210BCR/ABL-induced alteration of pre-mRNA splicing in primary human CD34+ hematopoietic progenitor cells. Leukemia 2004, 18, 727–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Kantarjian, H.; Zhang, X.; Wang, X.; Zhang, Z.; Yeh, C.-H.; O’Brien, S.; Giles, F.; Bruey, J.M.; Albitar, M. JAK2 Exon 14 Deletion in Patients with Chronic Myeloproliferative Neoplasms. PLoS ONE 2010, 5, e12165. [Google Scholar] [CrossRef] [Green Version]
- Catarsi, P.; Rosti, V.; Morreale, G.; Poletto, V.; Villani, L.; Bertorelli, R.; Pedrazzini, M.; Zorzetto, M.; Barosi, G. AGIMM Investigators JAK2 Exon 14 Skipping in Patients with Primary Myelofibrosis: A Minor Splice Variant Modulated by the JAK2-V617F Allele Burden. PLoS ONE 2015, 10, e0116636. [Google Scholar] [CrossRef]
- Nauroy, P.; Delhommeau, F.; Baklouti, F. JAK2V617F mRNA metabolism in myeloproliferative neoplasm cell lines. Blood Cancer J. 2014, 4, e222. [Google Scholar] [CrossRef] [Green Version]
- Fütterer, A.; Campanero, M.; Leonardo, E.; Criado, L.M.; Flores, J.M.; Hernández, J.M.; Miguel, J.F.S.; Martinez-A, C. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J. Clin. Investig. 2005, 115, 2351–2362. [Google Scholar] [CrossRef] [Green Version]
- Trachana, V.; Van Wely, K.H.M.; Guerrero, A.A.; Fütterer, A.; Martínez-A, C. Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proc. Natl. Acad. Sci. USA 2007, 104, 2691–2696. [Google Scholar] [CrossRef] [Green Version]
- Berzoti-Coelho, M.G.; Ferreira, A.F.; Nunes, N.D.S.; Pinto, M.; Júnior, M.C.R.; Simões, B.P.; Martinez-A, C.; Souto, E.X.; Panepucci, R.A.; Covas, D.T.; et al. The expression of Death Inducer-Obliterator (DIDO) variants in Myeloproliferative Neoplasms. Blood Cells Mol. Dis. 2016, 59, 25–30. [Google Scholar] [CrossRef]
- García-Domingo, D.; Ramírez, D.; De Buitrago, G.G.; Martinez-A, C. Death Inducer-Obliterator 1 Triggers Apoptosis after Nuclear Translocation and Caspase Upregulation. Mol. Cell. Boil. 2003, 23, 3216–3225. [Google Scholar] [CrossRef] [Green Version]
- Fütterer, A.; De Celis, J.; Navajas, R.; Almonacid, L.; Gutiérrez, J.; Talavera-Gutiérrez, A.; Pacios-Bras, C.; Bernascone, I.; Martin-Belmonte, F.; Martinéz-A, C. DIDO as a Switchboard that Regulates Self-Renewal and Differentiation in Embryonic Stem Cells. Stem Cell Rep. 2017, 8, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sabath, D.F.; Kuter, D.J. Cloning and Functional Characterization of a Novel C-Mpl Variant Expressed in Human Cd34 Cells and Platelets. Cytokine 2000, 12, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Skoda, R.; Seldin, D.; Chiang, M.; Peichel, C.L.; Vogt, T.; Leder, P. Murine c-mpl: A member of the hematopoietic growth factor receptor superfamily that transduces a proliferative signal. EMBO J. 1993, 12, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Mignotte, V.; Vigon, I.; De Crèvecoeur, E.B.; Romeo, P.-H.; Lemarchandel, V.; Chretien, S. Structure and Transcription of the Human c-mpl Gene (MPL). Genomes 1994, 20, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.-J.; ElKassar, N.; Hétet, G.; Brière, J.; Grandchamp, B.; Gardin, C. Study of the thrombopoietin receptor in essential thrombocythemia. Leukemia 1997, 11, 1821–1826. [Google Scholar] [CrossRef] [Green Version]
- Millot, G. MplK, a natural variant of the thrombopoietin receptor with a truncated cytoplasmic domain, binds thrombopoietin but does not interfere with thrombopoietin-mediated cell growth. Exp. Hematol. 2002, 30, 166–175. [Google Scholar] [CrossRef]
- Coers, J.; Ranft, C.; Skoda, R.C. A Truncated Isoform of c-Mpl with an Essential C-terminal Peptide Targets the Full-length Receptor for Degradation. J. Boil. Chem. 2004, 279, 36397–36404. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Sun, R.; Wu, L.; Huang, J.; Wang, P.; Yuan, H.; Qiu, F.; Xu, X.; Wu, D.; Yu, Y.; et al. Identification and characterization of an alternative splice variant of Mpl with a high affinity for TPO and its activation of ERK1/2 signaling. Int. J. Biochem. Cell Biol. 2013, 45, 2852–2863. [Google Scholar] [CrossRef]
- Chen, L.; Kostadima, M.A.; Martens, J.H.A.; Canu, G.; Garcia, S.P.; Turro, E.; Downes, K.; Macaulay, I.C.; Bielczyk-Maczynska, E.; Coe, S.; et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 2014, 345, 1251033. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.Y.-T.; Simon, L.M.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.V.; Sun, T.; Kurley, S.J.; Tyagi, S.; et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef]
- Theophile, K.; Buesche, G.; Kreipe, H.; Bock, O. The expression levels of telomerase catalytic subunit hTERT and oncogenic MYC in essential thrombocythemia are affected by the molecular subtype. Ann. Hematol. 2007, 87, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi-Tago, M.; Sumi, K.; Kasahara, T.; Tago, K. Critical Roles of Myc-ODC Axis in the Cellular Transformation Induced by Myeloproliferative Neoplasm-Associated JAK2 V617F Mutant. PLoS ONE 2013, 8, e52844. [Google Scholar] [CrossRef] [PubMed]
- Amirkhah, R.; Naderi-Meshkin, H.; Shah, J.S.; Dunne, P.D.; Schmitz, U. The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer. Cells 2019, 8, 929. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Pinello, N.; Nguyen, T.V.; Thoeng, A.; Nagarajah, R.; Holst, J.; Rasko, J.E.J.; Wong, J.J.-L. DNA methylation/hydroxymethylation regulate gene expression and alternative splicing during terminal granulopoiesis. Epigenomics 2019, 11, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodríguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Xiao, N.; Laha, S.; Das, S.P.; Morlock, K.; Jesneck, J.L.; Raffel, G.D. Ott1 (Rbm15) regulates thrombopoietin response in hematopoietic stem cells through alternative splicing of c-Mpl. Blood 2015, 125, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.-W.; Micol, J.-B.; Zhang, X.J.; De Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 4743. [Google Scholar] [CrossRef]
- Jiang, Q.; Crews, L.; Barrett, C.L.; Court-Recart, A.; Goff, D.; Sadarangani, A.; Rusert, J.; Morris, S.; Goldstein, L.; Chun, H.-J.; et al. Abstract 247: ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Tumor Biol. 2013, 73, 247. [Google Scholar] [CrossRef]
- Kędzierska, H.; Piekiełko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Metz, P.J.; Ching, K.A.; Xie, T.; Cuenca, P.D.; Niessen, S.; Tatlock, J.H.; Jensen-Pergakes, K.; Murray, B.W. Symmetric Arginine Dimethylation Is Selectively Required for mRNA Splicing and the Initiation of Type I and Type III Interferon Signaling. Cell Rep. 2020, 30, 1935–1950.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.C.-W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, W.; Yuan, Y. Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. J. Med. Chem. 2018, 61, 9429–9441. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bownan, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 inhibition modulates E2F1 methylation and gene regulatory networks leading to therapeutic efficacy in JAK2V617F mutant MPN. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef]
- Zhang, L.; Tran, N.-T.; Su, H.; Wang, R.; Lu, Y.; Tang, H.; Aoyagi, S.; Guo, A.; Khodadadi-Jamayran, A.; Zhou, D.; et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 2015, 4. [Google Scholar] [CrossRef]
- Shia, W.-J.; Okumura, A.J.; Yan, M.; Sarkeshik, A.; Lo, M.-C.; Matsuura, S.; Komeno, Y.; Zhao, X.; Nimer, S.D.; Yates, J.R.; et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 2012, 119, 4953–4962. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; He, X.; Lin, Y.-C.; Dong, H.; Zhang, L.; Chen, X.; Wang, Z.; Shen, Y.; Li, M.; Wang, H.; et al. Targeting PRMT1-mediated FLT3 methylation disrupts maintenance of MLL-rearranged acute lymphoblastic leukemia. Blood 2019, 134, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Bressan, G.C.; Moraes, E.C.; Manfiolli, A.O.; Kuniyoshi, T.M.; Pasos, D.O.; Gomes, M.D.; Kobarg, J. Arginine methylation analysis of the splicing-associated SR protein SFRS9/SRP30C. Cell. Mol. Biol. Lett. 2009, 14, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Stewart, C.; Abdel-Wahab, O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov. 2019, 9, 1493–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellagatti, A.; Boultwood, J. Splicing factor mutant myelodysplastic syndromes: Recent advances. Adv. Boil. Regul. 2020, 75, 100655. [Google Scholar] [CrossRef]
- Lee, S.C.-W.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.-B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.A.; Agrawal, A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Salton, M.; Kasprzak, W.K.; Voss, T.; Shapiro, B.A.; Poulikakos, P.I.; Misteli, T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Han, C.; Wang, E.; Lorch, A.H.; Serafin, V.; Cho, B.-K.; Diaz, B.T.G.; Calvo, J.; Fang, C.; Khodadadi-Jamayran, A.; et al. Posttranslational regulation of the exon skipping machinery controls aberrant splicing in leukemia. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Itskovich, S.S.; Gurunathan, A.; Clark, J.; Burwinkel, M.; Wunderlich, M.; Berger, M.R.; Kulkarni, A.; Chetal, K.; Venkatasubramanian, M.; Salomonis, N.; et al. MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Lee, S.C.-W.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]



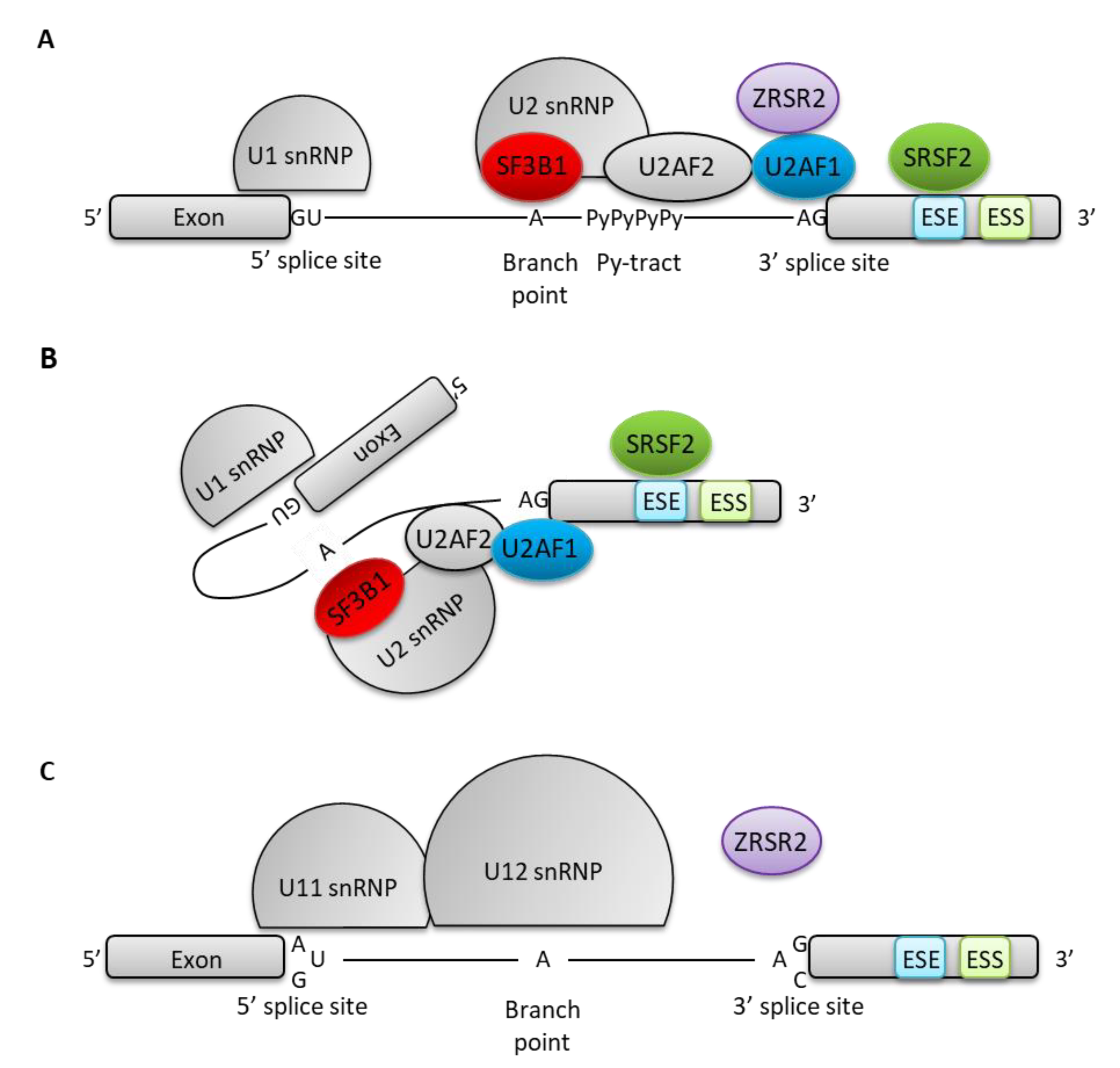{kind=link}
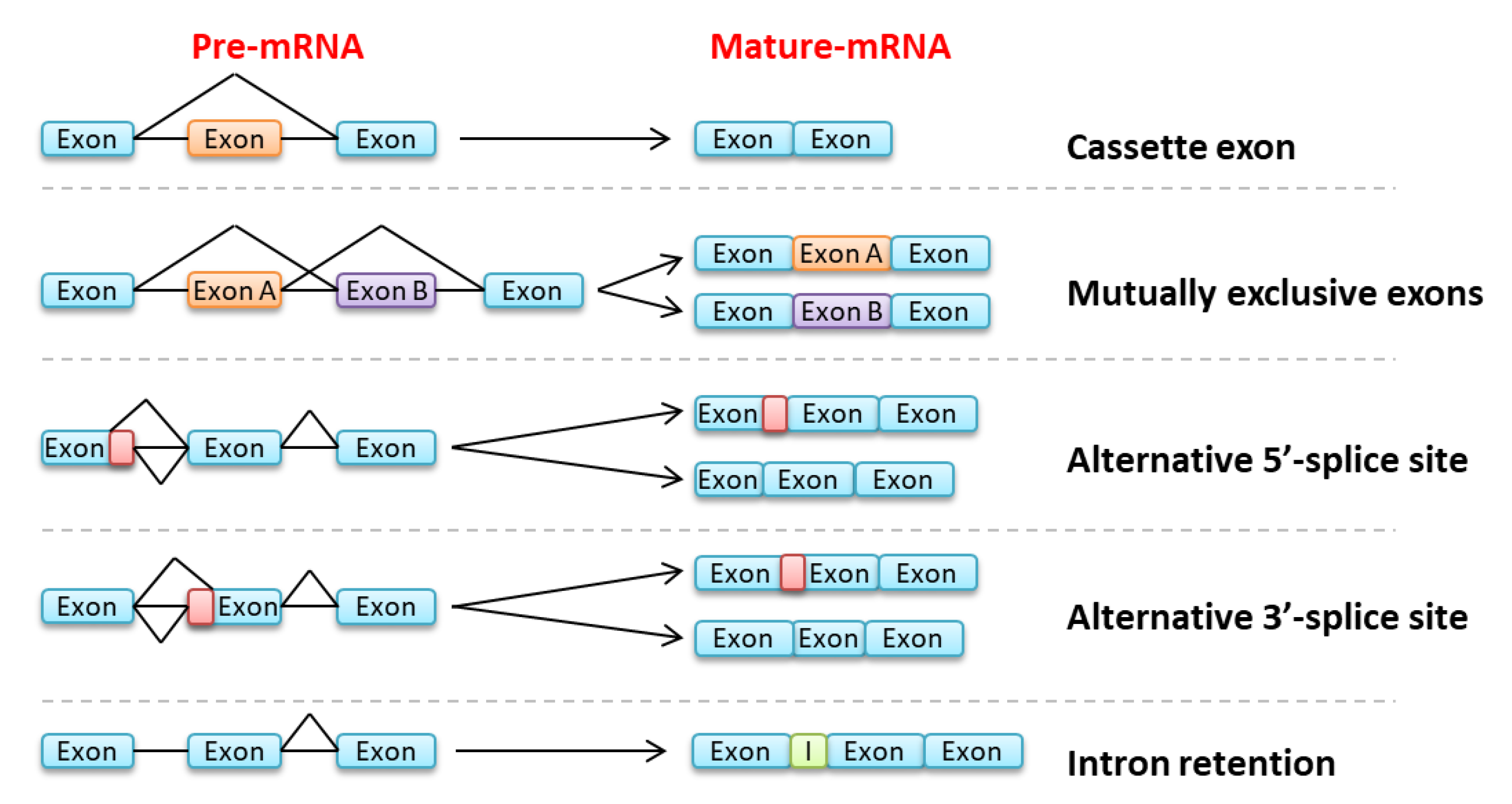{kind=link}
| Gene | Mutation Frequency (n/N) | References | ||||
|---|---|---|---|---|---|---|
| PMF | SMF | ET | PV | AML Post MPN | ||
| SRSF2 | 12.2% (351/2876) Pre-MF: 8.5% (25/294) | 2.6% (12/461) | 1.5% (31/2132) | 1.8% (22/1244) | 17% (25/147) | [24,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61] |
| SF3B1 | 7.7% (135/1751) | 6.9% (8/116) | 2.8% (62/2207) | 1.4% (14/1027) | 8.1% (13/161) | [45,46,47,48,49,50,51,53,55,56,57,58,59,60,61,62,63,64] |
| U2AF1 | 13.8% (200/1448) | 4.9% (5/102) | 1.1% (23/2132) | 0.3% (3/969) | 9.5% (14/147) | [46,47,48,49,50,53,55,56,57,58,59,60,61] |
| ZRSR2 | 5.8% (52/891) | 6.9% (7/102) | 0.7% (11/1590) | 1.3% (7/535) | 1.1% (1/91) | [46,47,48,55,56,57,58,59,61] |
| PRPF8 | 2.1% (2/94) | 1.6% (1/64) | 2.9% (1/35) | 0% (0/8) | 5.3% (5/94) | [48,50,59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hautin, M.; Mornet, C.; Chauveau, A.; Bernard, D.G.; Corcos, L.; Lippert, E. Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers 2020, 12, 2216. https://doi.org/10.3390/cancers12082216
Hautin M, Mornet C, Chauveau A, Bernard DG, Corcos L, Lippert E. Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers. 2020; 12(8):2216. https://doi.org/10.3390/cancers12082216
Chicago/Turabian StyleHautin, Marie, Clélia Mornet, Aurélie Chauveau, Delphine G. Bernard, Laurent Corcos, and Eric Lippert. 2020. "Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues" Cancers 12, no. 8: 2216. https://doi.org/10.3390/cancers12082216
APA StyleHautin, M., Mornet, C., Chauveau, A., Bernard, D. G., Corcos, L., & Lippert, E. (2020). Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers, 12(8), 2216. https://doi.org/10.3390/cancers12082216