Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms


{kind=link}
{kind=link}
Abstract
:1. Background
2. Inflammatory Cytokines and Their Sources in MPN
3. Allogeneic HCT: Conditioning, Transplantation, and Post-HCT Management
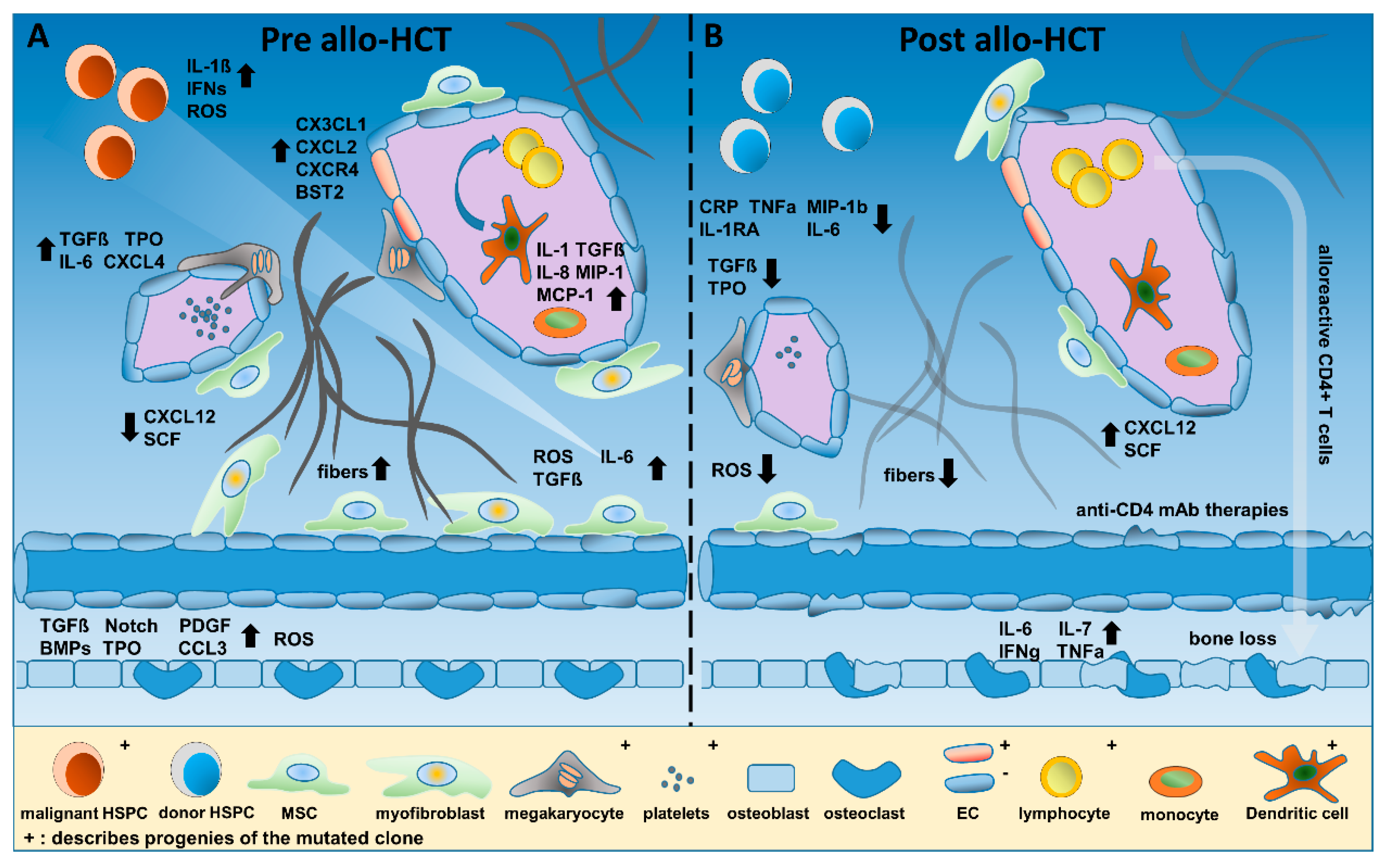
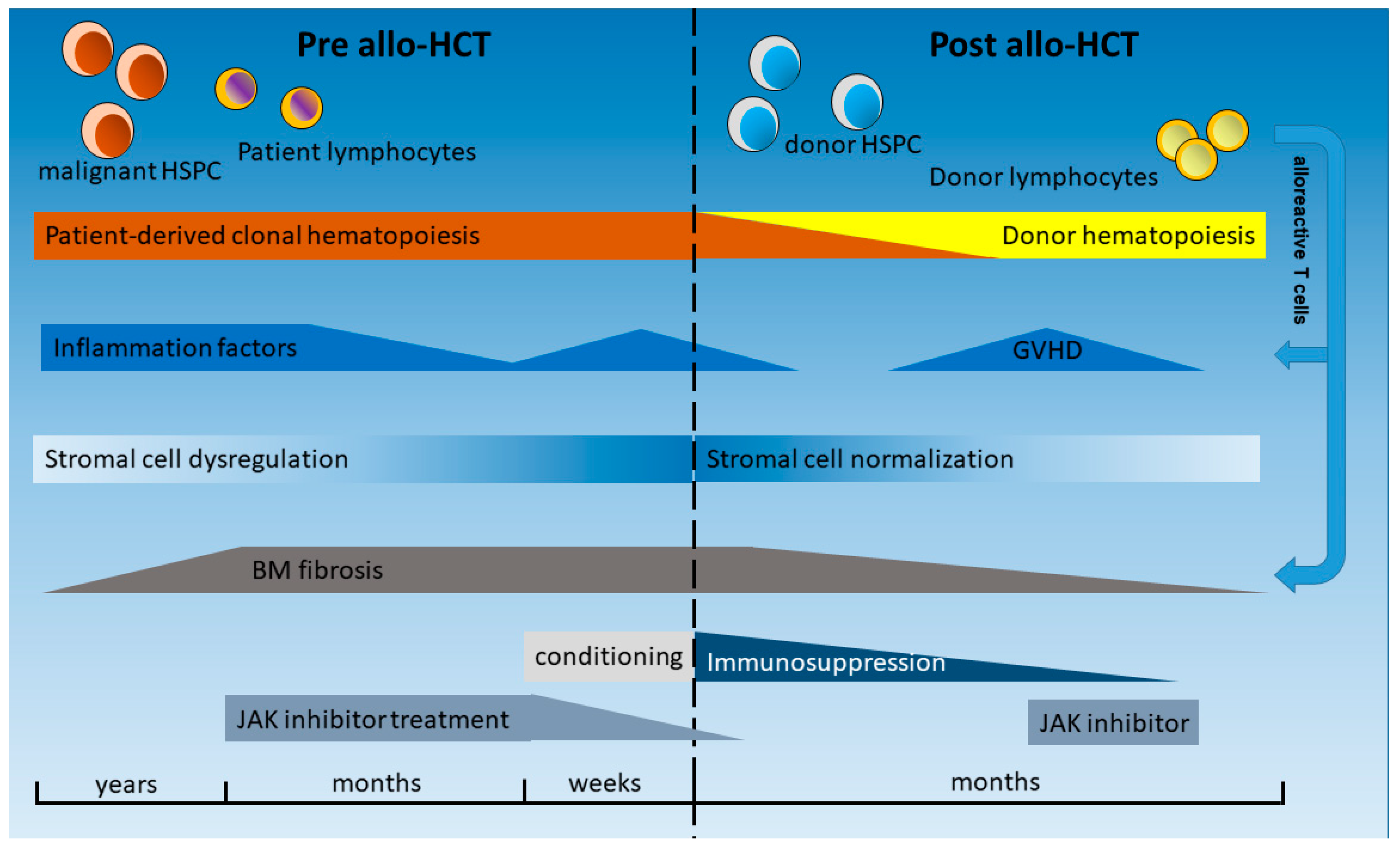
4. Inflammatory Conditions in the Hematopoietic Niche before and after HCT
5. Therapeutic Approaches to Reduce Inflammation and to Prevent HCT Graft Failure
5.1. JAK Inhibitors in HCT
5.2. Targeting ROS and Iron Overload
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of Myeloproliferative Disorders. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 101–126. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs. JAK2 vs. MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langabeer, S.E. Chasing down the triple-negative myeloproliferative neoplasms: Implications for molecular diagnostics. JAK-STAT 2016, 5, e1248011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, M.M.J.; Schemionek, M.; Schubert, C.; Chatain, N.; Sontag, S.; Isfort, S.; Ortiz-Brüchle, N.; Schmitt, K.; Krüger, L.; Zerres, K.; et al. Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS ONE 2015, 10, e0123476. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediat. Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef] [Green Version]
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020, 100711, in press. [Google Scholar]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, T.; Müller, M.; Stockinger, S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Schubert, C.; Köhler, J.; Schemionek, M.; Isfort, S.; Brümmendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Mendez Luque, L.F.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeg, H.J.; Gooley, T.A.; Flowers, M.E.D.; Sale, G.E.; Slattery, J.T.; Anasetti, C.; Chauncey, T.R.; Doney, K.; Georges, G.E.; Kiem, H.-P.; et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003, 102, 3912–3918. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J.; Appelbaum, F.R. Stem-cell transplantation for myelofibrosis. N. Engl. J. Med. 2001, 344, 775–776. [Google Scholar] [CrossRef]
- Guardiola, P.; Anderson, J.E.; Bandini, G.; Cervantes, F.; Runde, V.; Arcese, W.; Bacigalupo, A.; Przepiorka, D.; O’Donnell, M.R.; Polchi, P.; et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: A European Group for Blood and Marrow Transplantation, Société Française de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Ce. Blood 1999, 93, 2831–2838. [Google Scholar] [PubMed]
- Anderson, J.E.; Sale, G.; Appelbaum, F.R.; Chauncey, T.R.; Storb, R. Allogeneic marrow transplantation for primary myelofibrosis and myelofibrosis secondary to polycythaemia vera or essential thrombocytosis. Br. J. Haematol. 1997, 98, 1010–1016. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Mein, P.; Müller, C. ZKRD Zentrales Knochenmarkspender-Register Deutschland German Standards for Unrelated Blood Stem Cell Donations. Available online: https//www.ebmt.org/regulations-guidelines (accessed on 28 June 2020).
- Eva, L.; Grießhammer, M.; Petrides, P.E. Myeloproliferative Neoplasien (MPN) (Früher: Chronische Myeloproliferative Erkrankungen (CMPE)). Available online: https//www.onkopedia.com/de/onkopedia/guidelines/myeloproliferative-neoplasien-mpn-frueher-chronische-myeloproliferative-erkrankungen-cmpe/@@guideline/html/index.html (accessed on 28 June 2020).
- Kröger, N.; Holler, E.; Kobbe, G.; Bornhäuser, M.; Schwerdtfeger, R.; Baurmann, H.; Nagler, A.; Bethge, W.; Stelljes, M.; Uharek, L.; et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: A prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009, 114, 5264–5270. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. How I treat myelofibrosis. Blood 2011, 117, 3494–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, F. How I Treat. Blood 2014, 124, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Derlin, T.; Alchalby, H.; Bannas, P.; Veldhoen, S.; Apostolova, I.; Triviai, I.; Bengel, F.M.; Kröger, N. Assessment of bone marrow inflammation in patients with myelofibrosis: An 18F-fluorodeoxyglucose PET/CT study. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 696–705. [Google Scholar] [CrossRef]
- Ojeda-Uribe, M.; Morel, O.; Ungureanu, C.; Desterke, C.; Le Bousse-Kerdilès, M.-C.; Boulahdour, H. Assessment of sites of marrow and extramedullary hematopoiesis by hybrid imaging in primary myelofibrosis patients. Cancer Med. 2016, 5, 2378–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef]
- Ciaffoni, F.; Cassella, E.; Varricchio, L.; Massa, M.; Barosi, G.; Migliaccio, A.R. Activation of non-canonical TGF-β1 signaling indicates an autoimmune mechanism for bone marrow fibrosis in primary myelofibrosis. Blood Cells. Mol. Dis. 2015, 54, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Čokić, V.P.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.B.; Marković, D.; Buač, M.; Diklić, M.; Kraguljac-Kurtović, N.; Damjanović, S.; Milenković, P.; Gotić, M.; et al. Proinflammatory Cytokine IL-6 and JAK-STAT Signaling Pathway in Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 453020. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Heaton, W.L.; Senina, A.V.; Pomicter, A.D.; Salama, M.E.; Clair, P.M.; Yan, D.; Bell, R.N.; Gililland, J.M.; Prchal, J.T.; O’Hare, T.; et al. Autocrine Tnf signaling favors malignant cells in myelofibrosis in a Tnfr2-dependent fashion. Leukemia 2018, 32, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Alonci, A.; Bellomo, G.; Campo, S.; Cannavò, A.; Penna, G.; Russo, S.; Centorrino, R.; Gerace, D.; Petrungaro, A.; et al. Increased serum levels of neutrophil gelatinase-associated lipocalin in patients with essential thrombocythemia and polycythemia vera. Leuk. Lymphoma 2011, 52, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Xia, L.; Liu, Y.-C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramann, R.; Schneider, R.K. The identification of fibrosis-driving myofibroblast precursors reveals new therapeutic avenues in myelofibrosis. Blood 2018, 131, 2111–2119. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800.e8. [Google Scholar] [CrossRef] [Green Version]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C.; Larsen, T.S. Gene expression profiling with principal component analysis depicts the biological continuum from essential thrombocythemia over polycythemia vera to myelofibrosis. Exp. Hematol. 2012, 40, 771–780.e19. [Google Scholar] [CrossRef]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maié, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Schubert, C.; Allhoff, M.; Tillmann, S.; Maié, T.; Costa, I.G.; Lipka, D.B.; Schemionek, M.; Feldberg, K.; Baumeister, J.; Brümmendorf, T.H.; et al. Differential roles of STAT1 and STAT2 in the sensitivity of JAK2V617F- vs. BCR-ABL-positive cells to interferon alpha. J. Hematol. Oncol. 2019, 12, 36. [Google Scholar] [CrossRef]
- Chen, E.; Beer, P.A.; Godfrey, A.L.; Ortmann, C.A.; Li, J.; Costa-Pereira, A.P.; Ingle, C.E.; Dermitzakis, E.T.; Campbell, P.J.; Green, A.R. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell 2010, 18, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.J.; Baltay, M.; Getz, A.; Fuhrman, K.; Aster, J.C.; Hasserjian, R.P.; Pozdnyakova, O. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLoS ONE 2019, 14, e0216810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, P.; Palucka, A.; Pascual, V.; Banchereau, J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology 2017, 6, e1345402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humblet-Baron, S.; Barber, J.S.; Roca, C.P.; Lenaerts, A.; Koni, P.A.; Liston, A. Murine myeloproliferative disorder as a consequence of impaired collaboration between dendritic cells and CD4 T cells. Blood 2019, 133, 319–330. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Merlinsky, T.R.; Levine, R.L.; Pronier, E. Unfolding the Role of Calreticulin in Myeloproliferative Neoplasm Pathogenesis. Clin. Cancer Res. 2019, 25, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2(V617F) myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, J.; Chatain, N.; Hubrich, A.; Maié, T.; Costa, I.G.; Denecke, B.; Han, L.; Küstermann, C.; Sontag, S.; Seré, K.; et al. Hypoxia-inducible factor 1 (HIF-1) is a new therapeutic target in JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2020, 34, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Mendez Luque, L.F.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Asenova, S.; Badbaran, A.; Zander, A.R.; Alchalby, H.; Fehse, B.; Kröger, N.; Lange, C.; Ayuk, F. Bone marrow mesenchymal stromal cells remain of recipient origin after allogeneic SCT and do not harbor the JAK2V617F mutation in patients with myelofibrosis. Clin. Exp. Med. 2010, 10, 205–208. [Google Scholar] [CrossRef]
- Von der Heide, E.K.; Neumann, M.; Vosberg, S.; James, A.R.; Schroeder, M.P.; Ortiz-Tanchez, J.; Isaakidis, K.; Schlee, C.; Luther, M.; Jöhrens, K.; et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2017, 31, 1069–1078. [Google Scholar] [CrossRef]
- Kim, Y.; Jekarl, D.W.; Kim, J.; Kwon, A.; Choi, H.; Lee, S.; Kim, Y.-J.; Kim, H.-J.; Kim, Y.; Oh, I.-H.; et al. Genetic and epigenetic alterations of bone marrow stromal cells in myelodysplastic syndrome and acute myeloid leukemia patients. Stem Cell Res. 2015, 14, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Kuter, D.J.; Bain, B.; Mufti, G.; Bagg, A.; Hasserjian, R.P. Bone marrow fibrosis: Pathophysiology and clinical significance of increased bone marrow stromal fibres. Br. J. Haematol. 2007, 139, 351–362. [Google Scholar] [CrossRef]
- Byrne, J.L.; Beshti, H.; Clark, D.; Ellis, I.; Haynes, A.P.; Das-Gupta, E.; Russell, N.H. Induction of remission after donor leucocyte infusion for the treatment of relapsed chronic idiopathic myelofibrosis following allogeneic transplantation: Evidence for a “graft vs. myelofibrosis” effect. Br. J. Haematol. 2000, 108, 430–433. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Diehl, V. Standardization of bone marrow features—Does it work in hematopathology for histological discrimination of different disease patterns? Histol. Histopathol. 2005, 20, 633–644. [Google Scholar]
- Kröger, N.; Zabelina, T.; Alchalby, H.; Stübig, T.; Wolschke, C.; Ayuk, F.; von Hünerbein, N.; Kvasnicka, H.-M.; Thiele, J.; Kreipe, H.-H.; et al. Dynamic of bone marrow fibrosis regression predicts survival after allogeneic stem cell transplantation for myelofibrosis. Biol. Blood Marrow Transplant. 2014, 20, 812–815. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, F.; Rovira, M.; Urbano-Ispizua, A.; Rozman, M.; Carreras, E.; Montserrat, E. Complete remission of idiopathic myelofibrosis following donor lymphocyte infusion after failure of allogeneic transplantation: Demonstration of a graft-versus-myelofibrosis effect. Bone Marrow Transplant. 2000, 26, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Klyuchnikov, E.; Holler, E.; Bornhäuser, M.; Kobbe, G.; Nagler, A.; Shimoni, A.; Könecke, C.; Wolschke, C.; Bacher, U.; Zander, A.R.; et al. Donor lymphocyte infusions and second transplantation as salvage treatment for relapsed myelofibrosis after reduced-intensity allografting. Br. J. Haematol. 2012, 159, 172–181. [Google Scholar] [CrossRef]
- Parampalli Yajnanarayana, S.; Stübig, T.; Cornez, I.; Alchalby, H.; Schönberg, K.; Rudolph, J.; Triviai, I.; Wolschke, C.; Heine, A.; Brossart, P.; et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br. J. Haematol. 2015, 169, 824–833. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin. Proc. 2011, 86, 1188–1191. [Google Scholar] [CrossRef]
- Carreras, E.; Diaz-Ricart, M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant. 2011, 46, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef]
- Ho, Y.-H.; Del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; García-García, A.; Macías, D.; González-Gómez, C.; Del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418.e6. [Google Scholar] [CrossRef] [Green Version]
- Galán-Díez, M.; Kousteni, S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr. Mol. Biol. Rep. 2017, 3, 53–62. [Google Scholar] [CrossRef]
- Moore, S.G.; Dawson, K.L. Red and yellow marrow in the femur: Age-related changes in appearance at MR imaging. Radiology 1990, 175, 219–223. [Google Scholar] [CrossRef]
- Tavassoli, M.; Crosby, W.H. Bone marrow histogenesis: A comparison of fatty and red marrow. Science 1970, 169, 291–293. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Laperrousaz, B.; Jeanpierre, S.; Sagorny, K.; Voeltzel, T.; Ramas, S.; Kaniewski, B.; Ffrench, M.; Salesse, S.; Nicolini, F.E.; Maguer-Satta, V. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood 2013, 122, 3767–3777. [Google Scholar] [CrossRef] [Green Version]
- McClune, B.L.; Majhail, N.S. Osteoporosis after stem cell transplantation. Curr. Osteoporos. Rep. 2013, 11, 305–310. [Google Scholar] [CrossRef]
- Tauchmanovaà, L.; Colao, A.; Lombardi, G.; Rotoli, B.; Selleri, C. REVIEW: Bone Loss and Its Management in Long-Term Survivors from Allogeneic Stem Cell Transplantation. J. Clin. Endocrinol. Metab. 2007, 92, 4536–4545. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-Y.; Baek, K.-H.; Rhee, E.-J.; Tae, H.-J.; Oh, K.-W.; Kang, M.-I.; Lee, K.-W.; Kim, S.-W.; Kim, C.-C.; Oh, E.-S. Impact of circulating bone-resorbing cytokines on the subsequent bone loss following bone marrow transplantation. Bone Marrow Transplant. 2004, 34, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Shono, Y.; Ueha, S.; Wang, Y.; Abe, J.; Kurachi, M.; Matsuno, Y.; Sugiyama, T.; Nagasawa, T.; Imamura, M.; Matsushima, K. Bone marrow graft-versus-host disease: Early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood 2010, 115, 5401–5411. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Iachininoto, M.G.; Capodimonti, S.; Nuzzolo, E.R.; Torti, L.; Cenci, T.; Larocca, L.M.; Leone, G. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 2011, 117, 2700–2707. [Google Scholar] [CrossRef]
- Rosti, V.; Villani, L.; Riboni, R.; Poletto, V.; Bonetti, E.; Tozzi, L.; Bergamaschi, G.; Catarsi, P.; Dallera, E.; Novara, F.; et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 2013, 121, 360–368. [Google Scholar] [CrossRef]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]
- Guadall, A.; Lesteven, E.; Letort, G.; Awan Toor, S.; Delord, M.; Pognant, D.; Brusson, M.; Verger, E.; Maslah, N.; Giraudier, S.; et al. Endothelial Cells Harbouring the JAK2V617F Mutation Display Pro-Adherent and Pro-Thrombotic Features. Thromb. Haemost. 2018, 118, 1586–1599. [Google Scholar] [CrossRef] [Green Version]
- Etheridge, S.L.; Roh, M.E.; Cosgrove, M.E.; Sangkhae, V.; Fox, N.E.; Chen, J.; López, J.A.; Kaushansky, K.; Hitchcock, I.S. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, 2295–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushansky, K.; Zhan, H. The regulation of normal and neoplastic hematopoiesis is dependent on microenvironmental cells. Adv. Biol. Regul. 2018, 69, 11–15. [Google Scholar] [CrossRef]
- Guy, A.; Danaee, A.; Paschalaki, K.; Boureau, L.; Rivière, E.; Etienne, G.; Mansier, O.; Laffan, M.; Sekhar, M.; James, C. Absence of JAK2V617F Mutated Endothelial Colony-Forming Cells in Patients With JAK2V617F Myeloproliferative Neoplasms and Splanchnic Vein Thrombosis. HemaSphere 2020, 4, e364. [Google Scholar] [CrossRef]
- Thiele, J.; Varus, E.; Siebolts, U.; Kvasnicka, H.M.; Wickenhauser, C.; Metz, K.A.; Beelen, D.W.; Ditschkowski, M.; Zander, A.; Kröger, N. Dualism of mixed chimerism between hematopoiesis and stroma in chronic idiopathic myelofibrosis after allogeneic stem cell transplantation. Histol. Histopathol. 2007, 22, 365–372. [Google Scholar]
- Wong, K.M.; Atenafu, E.G.; Kim, D.; Kuruvilla, J.; Lipton, J.H.; Messner, H.; Gupta, V. Incidence and risk factors for early hepatotoxicity and its impact on survival in patients with myelofibrosis undergoing allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2012, 18, 1589–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; Kosiorek, H.E.; Mead, A.; Klisovic, R.B.; Galvin, J.P.; Berenzon, D.; Yacoub, A.; Viswabandya, A.; Mesa, R.A.; Goldberg, J.; et al. Ruxolitinib Therapy Followed by Reduced-Intensity Conditioning for Hematopoietic Cell Transplantation for Myelofibrosis: Myeloproliferative Disorders Research Consortium 114 Study. Biol. Blood Marrow Transplant. 2019, 25, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Boluda, J.C.; Pereira, A.; Kröger, N.; Beelen, D.; Robin, M.; Bornhäuser, M.; Angelucci, E.; Vitek, A.; Blau, I.W.; Niittyvuopio, R.; et al. Determinants of survival in myelofibrosis patients undergoing allogeneic hematopoietic cell transplantation. Leukemia 2020, 1–10. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Malcovati, L.; Boveri, E.; Travaglino, E.; Pietra, D.; Pascutto, C.; Passamonti, F.; Invernizzi, R.; Castello, A.; Magrini, U.; et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J. Clin. Oncol. 2009, 27, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Zabelina, T.; van Biezen, A.; Brand, R.; Niederwieser, D.; Martino, R.; Lim, Z.Y.; Onida, F.; Schmid, C.; Garderet, L.; et al. Allogeneic stem cell transplantation for myelodysplastic syndromes with bone marrow fibrosis. Haematologica 2011, 96, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, J.; Kvasnicka, H.M.; Dietrich, H.; Stein, G.; Hann, M.; Kaminski, A.; Rathjen, N.; Metz, K.A.; Beelen, D.W.; Ditschkowski, M.; et al. Dynamics of bone marrow changes in patients with chronic idiopathic myelofibrosis following allogeneic stem cell transplantation. Histol. Histopathol. 2005, 20, 879–889. [Google Scholar]
- Avanzini, M.A.; Bernardo, M.E.; Novara, F.; Mantelli, M.; Poletto, V.; Villani, L.; Lenta, E.; Ingo, D.M.; Achille, V.; Bonetti, E.; et al. Functional and genetic aberrations of in vitro-cultured marrow-derived mesenchymal stromal cells of patients with classical Philadelphia-negative myeloproliferative neoplasms. Leukemia 2014, 28, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Nagorney, D.S.; Schwager, S.; Allred, J.; Tefferi, A. Palliative goals, patient selection, and perioperative platelet management: Outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006, 107, 361–370. [Google Scholar] [CrossRef]
- Tanner, M.L.; Hoh, C.K.; Bashey, A.; Holman, P.; Sun, C.; Broome, H.E.; Lane, T.; Ball, E.D.; Carrier, E. FLAG chemotherapy followed by allogeneic stem cell transplant using nonmyeloablative conditioning induces regression of myelofibrosis with myeloid metaplasia. Bone Marrow Transplant. 2003, 32, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Hussein, K.; Stucki-Koch, A.; Alchalby, H.; Triviai, I.; Kröger, N.; Kreipe, H. Cytokine Expression Pattern in Bone Marrow Microenvironment after Allogeneic Stem Cell Transplantation in Primary Myelofibrosis. Biol. Blood Marrow Transplant. 2016, 22, 644–650. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.M.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef]
- Mengarelli, A.; Iori, A.; Guglielmi, C.; Romano, A.; Cerretti, R.; Torromeo, C.; Micozzi, A.; Fenu, S.; Laurenti, L.; Donato, V.; et al. Standard versus alternative myeloablative conditioning regimens in allogeneic hematopoietic stem cell transplantation for high-risk acute leukemia. Haematologica 2002, 87, 52–58. [Google Scholar]
- Kuss, B.J.; Sage, R.E.; Shepherd, K.M.; Hardingham, J.; Nicola, M. High dose hydroxyurea in collection of Philadelphia chromosome-negative stem cells in chronic myeloid leukaemia. Leuk. Lymphoma 1993, 10, 73–78. [Google Scholar] [CrossRef]
- Malato, A.; Rossi, E.; Tiribelli, M.; Mendicino, F.; Pugliese, N. Splenectomy in Myelofibrosis: Indications, Efficacy, and Complications. Clin. Lymphoma. Myeloma Leuk. 2020, 1–8. [Google Scholar] [CrossRef]
- Li, Z.; Deeg, H.J. Pros and cons of splenectomy in patients with myelofibrosis undergoing stem cell transplantation. Leukemia 2001, 15, 465–467. [Google Scholar] [CrossRef]
- Li, Z.; Gooley, T.; Appelbaum, F.R.; Deeg, H.J. Splenectomy and hemopoietic stem cell transplantation for myelofibrosis. Blood 2001, 97, 2180–2181. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef] [Green Version]
- Kvasnicka, H.M.; Thiele, J.; Bueso-Ramos, C.E.; Sun, W.; Cortes, J.; Kantarjian, H.M.; Verstovsek, S. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J. Hematol. Oncol. 2018, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- McLornan, D.P.; Yakoub-Agha, I.; Robin, M.; Chalandon, Y.; Harrison, C.N.; Kroger, N. State-of-the-art review: Allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica 2019, 104, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Stübig, T.; Alchalby, H.; Ditschkowski, M.; Wolf, D.; Wulf, G.; Zabelina, T.; Wolschke, C.; Ayuk, F.; Kröger, N. JAK inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post-ET/PV myelofibrosis. Leukemia 2014, 28, 1736–1738. [Google Scholar] [CrossRef]
- Schönberg, K.; Rudolph, J.; Vonnahme, M.; Parampalli Yajnanarayana, S.; Cornez, I.; Hejazi, M.; Manser, A.R.; Uhrberg, M.; Verbeek, W.; Koschmieder, S.; et al. JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Res. 2015, 75, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, S.; Tateno, T.; Ito, S.; Tsutsumi, Y.; Teshima, T. Evaluation of Short-Term Ruxolitinib Tapering Strategy Before Allogeneic Stem Cell Transplantation for Primary Myelofibrosis Through the Transition of Serum Cytokines and Growth Factors. Transplant. Direct 2016, 2, e95. [Google Scholar] [CrossRef]
- Kröger, N.M.; Deeg, J.H.; Olavarria, E.; Niederwieser, D.; Bacigalupo, A.; Barbui, T.; Rambaldi, A.; Mesa, R.; Tefferi, A.; Griesshammer, M.; et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: A consensus process by an EBMT/ELN international working group. Leukemia 2015, 29, 2126–2133. [Google Scholar] [CrossRef]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef]
- Carniti, C.; Gimondi, S.; Vendramin, A.; Recordati, C.; Confalonieri, D.; Bermema, A.; Corradini, P.; Mariotti, J. Pharmacologic Inhibition of JAK1/JAK2 Signaling Reduces Experimental Murine Acute GVHD While Preserving GVT Effects. Clin. Cancer Res. 2015, 21, 3740–3749. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, R.; Burchert, A.; Lengerke, C.; Verbeek, M.; Maas-Bauer, K.; Metzelder, S.K.; Spoerl, S.; Ditschkowski, M.; Ecsedi, M.; Sockel, K.; et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: A multicenter survey. Leukemia 2015, 29, 2062–2068. [Google Scholar] [CrossRef]
- Pfeifer, H.; Wassmann, B.; Bethge, W.; Dengler, J.; Bornhäuser, M.; Stadler, M.; Beelen, D.; Vucinic, V.; Burmeister, T.; Stelljes, M.; et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia 2013, 27, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Warraich, Z.; Tenneti, P.; Thai, T.; Hubben, A.; Amin, H.; McBride, A.; Warraich, S.; Hannan, A.; Warraich, F.; Majhail, N.; et al. Relapse Prevention with Tyrosine Kinase Inhibitors after Allogeneic Transplantation for Philadelphia Chromosome–Positive Acute Lymphoblast Leukemia: A Systematic Review. Biol. Blood Marrow Transplant. 2020, 26, e55–e64. [Google Scholar] [CrossRef]
- Fouillet, L.; Daguenet, E.; Schein, F.; Tavernier, E.; Flandrin-Gresta, P.; Cornillon, J. Clonal evolution of myelofibrosis treated with hematopoietic transplantation, using RUXOLITINIB for chronic GvHD: A case report. Curr. Res. Transl. Med. 2018, 66, 111–113. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, Y.; Zhang, Y.-Y.; Shi, M.-M.; Mo, X.-D.; Sun, Y.-Q.; Chang, Y.-J.; Xu, L.-P.; Zhang, X.-H.; Liu, K.-Y.; et al. Prophylactic oral NAC reduced poor hematopoietic reconstitution by improving endothelial cells after haploidentical transplantation. Blood Adv. 2019, 3, 1303–1317. [Google Scholar] [CrossRef]
- Cao, X.-N.; Kong, Y.; Song, Y.; Shi, M.-M.; Zhao, H.-Y.; Wen, Q.; Lyu, Z.-S.; Duan, C.-W.; Wang, Y.; Xu, L.-P.; et al. Impairment of bone marrow endothelial progenitor cells in acute graft-versus-host disease patients after allotransplant. Br. J. Haematol. 2018, 182, 870–886. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Suzuki, S.; Lai, C.-Y.; Yamazaki, S.; Kakuta, S.; Iwakura, Y.; Nojima, M.; Takeuchi, Y.; Higashihara, M.; Nakauchi, H.; et al. Pre-Transplantation Blockade of TNF-α-Mediated Oxygen Species Accumulation Protects Hematopoietic Stem Cells. Stem Cells 2017, 35, 989–1002. [Google Scholar] [CrossRef]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.-U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, O.; Kuepper, M.K.; Bütow, M.; Costa, I.G.; Appelmann, I.; Beier, F.; Luedde, T.; Braunschweig, T.; Koschmieder, S.; Brümmendorf, T.H.; et al. Infliximab therapy together with tyrosine kinase inhibition targets leukemic stem cells in chronic myeloid leukemia. BMC Cancer 2019, 19, 658. [Google Scholar] [CrossRef] [Green Version]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Ma, S.; Gong, H.; Liu, S.; Lei, L.; Hu, B.; Xu, Y.; Liu, H.; Wu, D. Inhibition of Acute Graft-versus-Host Disease with Retention of Graft-versus-Tumor Effects by Dimethyl Fumarate. Front. Immunol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Evens, A.M.; Mehta, J.; Gordon, L.I. Rust and corrosion in hematopoietic stem cell transplantation: The problem of iron and oxidative stress. Bone Marrow Transplant. 2004, 34, 561–571. [Google Scholar] [CrossRef]
- Pardanani, A.; Finke, C.; Abdelrahman, R.A.; Lasho, T.L.; Tefferi, A. Associations and prognostic interactions between circulating levels of hepcidin, ferritin and inflammatory cytokines in primary myelofibrosis. Am. J. Hematol. 2013, 88, 312–316. [Google Scholar] [CrossRef]
- Okabe, H.; Suzuki, T.; Uehara, E.; Ueda, M.; Nagai, T.; Ozawa, K. The bone marrow hematopoietic microenvironment is impaired in iron-overloaded mice. Eur. J. Haematol. 2014, 93, 118–128. [Google Scholar] [CrossRef]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 2015, 5, e10181. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, A.; Campagna, A.; De Muro, M.; Maurillo, L.; Trawinska, M.M.; LeonettiCrescenzi, S.; Petriccione, L.; Romano, A.; D’Addosio, A.; Cenfra, A.; et al. Deferasirox in the treatment of iron overload during myeloproliferative neoplasms in fibrotic phase: Does ferritin decrement matter? Leuk. Res. 2019, 76, 65–69. [Google Scholar] [CrossRef]
- Latagliata, R.; Montagna, C.; Porrini, R.; Di Veroli, A.; Leonetti, S.C.; Niscola, P.; Ciccone, F.; Spadea, A.; Breccia, M.; Maurillo, L.; et al. Chelation efficacy and erythroid response during deferasirox treatment in patients with myeloproliferative neoplasms in fibrotic phase. Eur. J. Haematol. 2016, 96, 643–649. [Google Scholar] [CrossRef]
- Maximova, N.; Gregori, M.; Simeone, R.; Sonzogni, A.; Boz, G.; Fucile, C.; Marini, V.; Martelli, A.; Mattioli, F. Safety and tolerability of deferasirox in pediatric hematopoietic stem cell transplant recipients: One facility’s five years’ experience of chelation treatment. Oncotarget 2017, 8, 63177–63186. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatain, N.; Koschmieder, S.; Jost, E. Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms. Cancers 2020, 12, 2250. https://doi.org/10.3390/cancers12082250
Chatain N, Koschmieder S, Jost E. Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms. Cancers. 2020; 12(8):2250. https://doi.org/10.3390/cancers12082250
Chicago/Turabian StyleChatain, Nicolas, Steffen Koschmieder, and Edgar Jost. 2020. "Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms" Cancers 12, no. 8: 2250. https://doi.org/10.3390/cancers12082250
APA StyleChatain, N., Koschmieder, S., & Jost, E. (2020). Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms. Cancers, 12(8), 2250. https://doi.org/10.3390/cancers12082250