A Biobank of Colorectal Cancer Patient-Derived Xenografts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
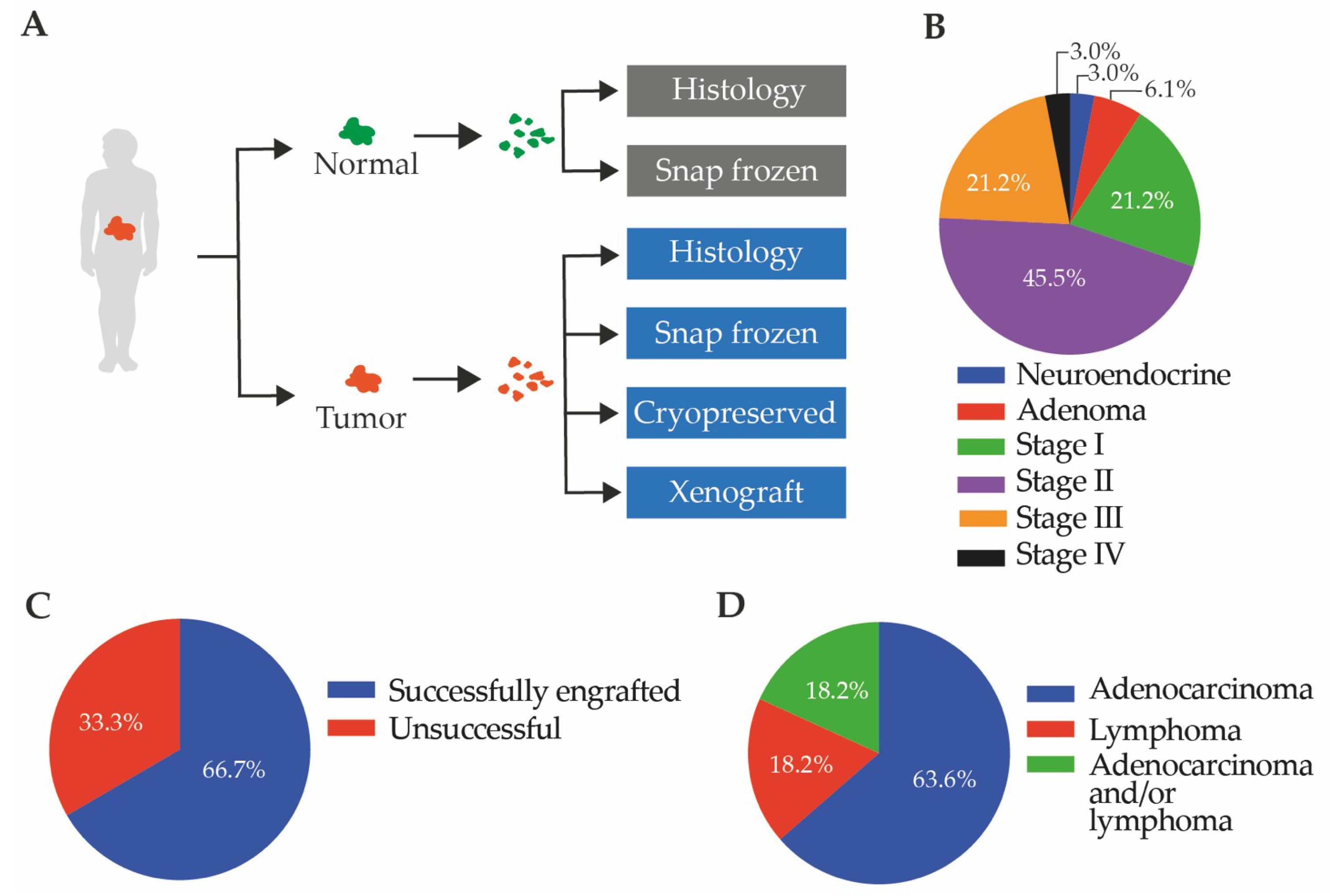
2.1. Establishment of Patient-Derived Xenografts
2.1.1. The Patient-Derived Xenografts Are Representative of Diverse Patient Populations
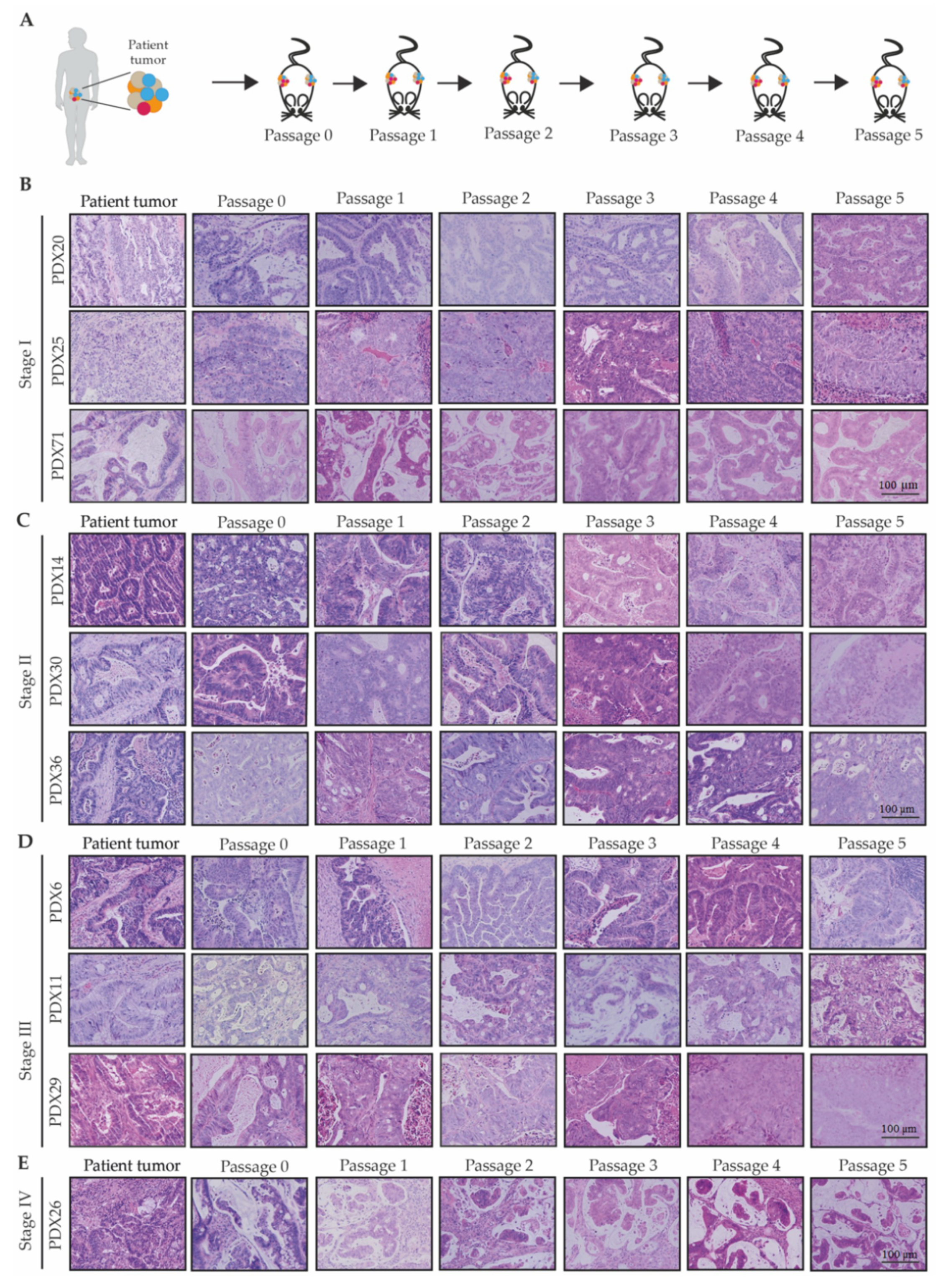
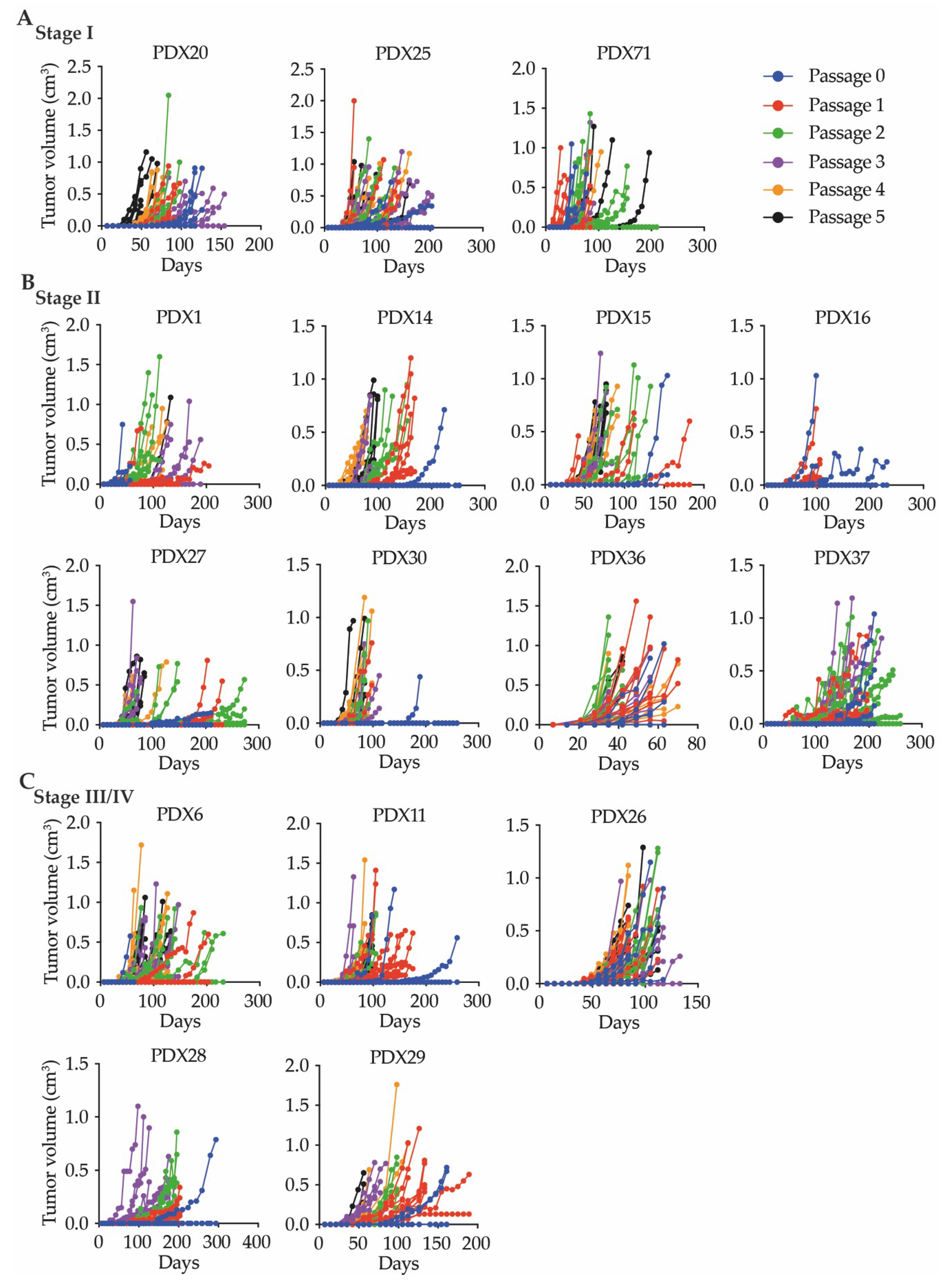
2.1.2. The CRC Patient-Derived Xenografts Successfully Underwent Serial Transplantation
2.1.3. The Patient-Derived Xenografts Maintained the Histological Features of the Patient Tumor
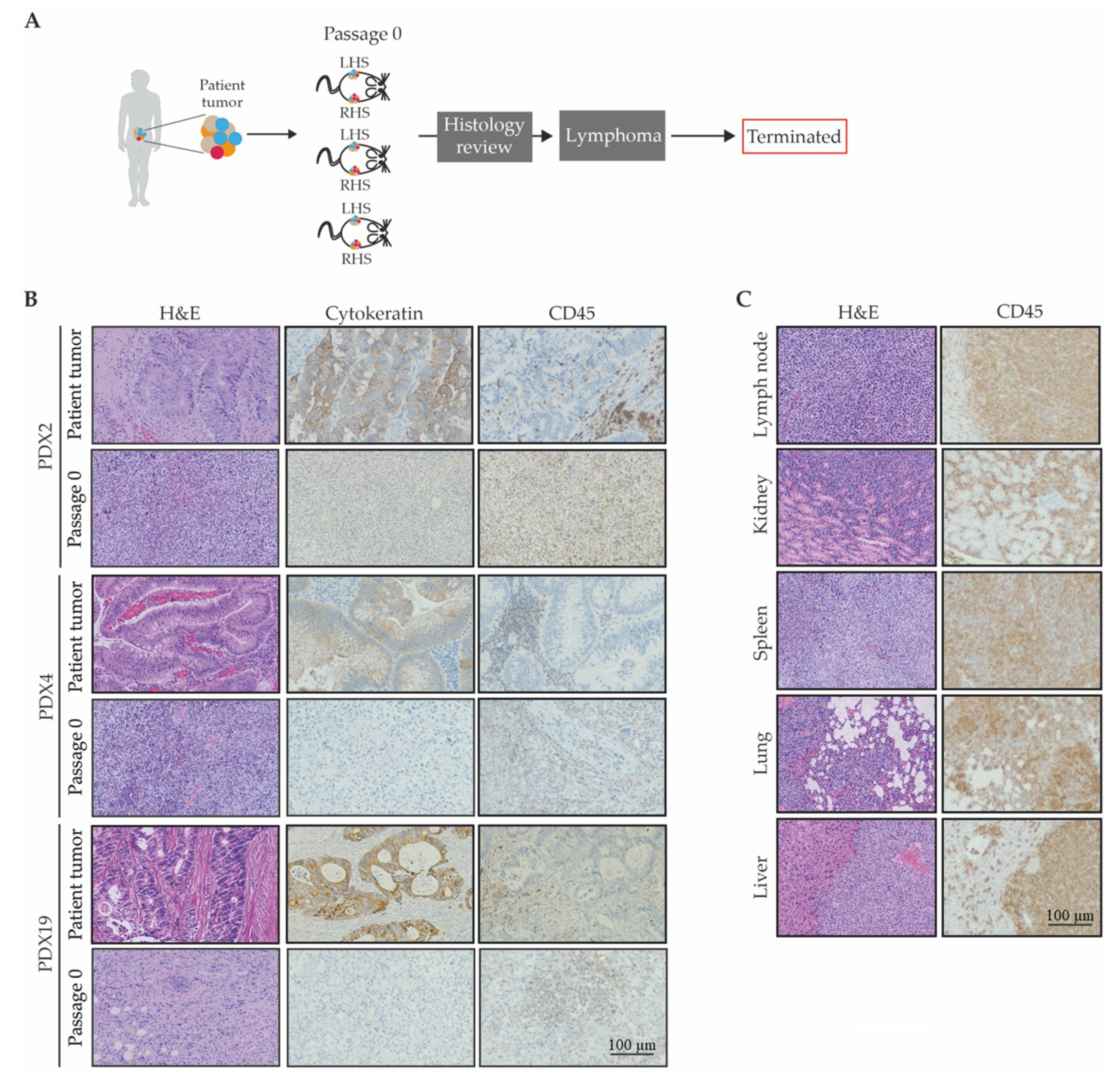
2.1.4. A Lymphocytic Phenotype Emerged for a Sub-Set of the Patient-Derived Xenografts
2.2. Characterization of Patient-Derived Xenografts
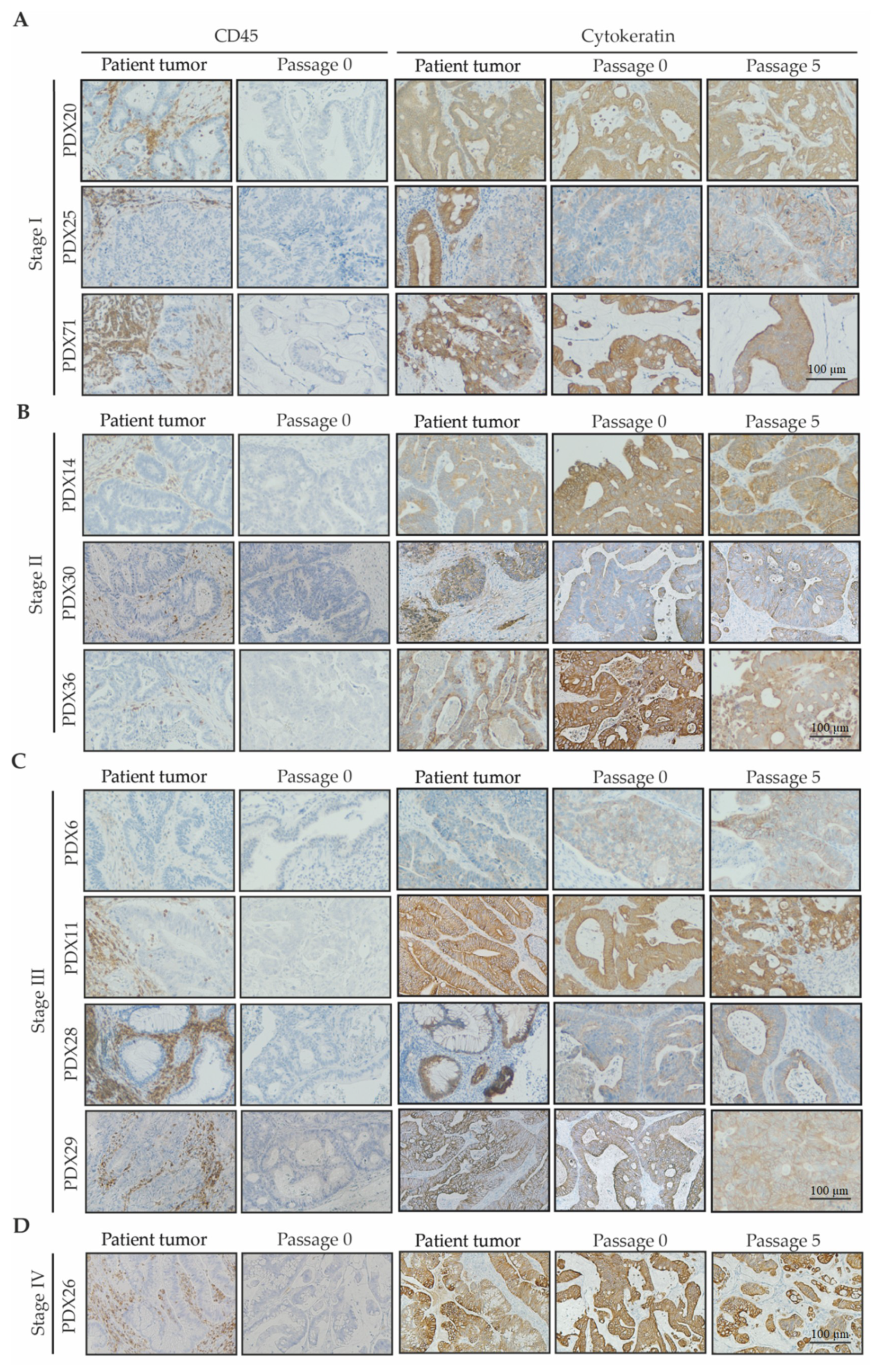
2.2.1. Human Stromal Cells Do Not Engraft with Patient Tumor Tissue
2.2.2. Genetic Signatures Are Retained in PDX Tumors during Early Serial Passages
2.2.3. Serial Passaging Increased the Successful Growth of Individual Tumors
2.3. Therapeutic Treatment of Patient-Derived Xenografts
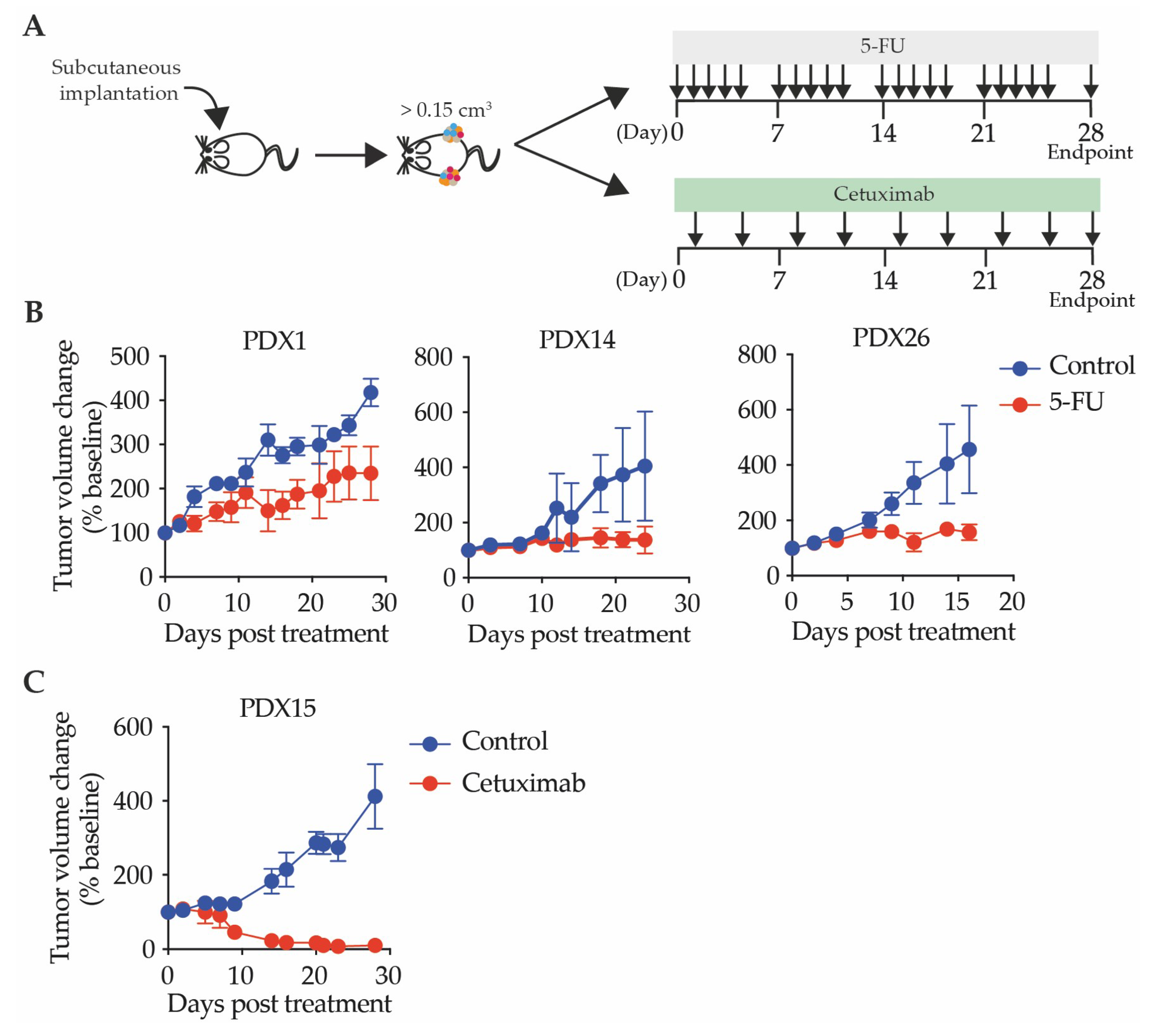
2.3.1. Patient-Derived Xenograft Response to Chemotherapy Reflects the Patient Response
2.3.2. Patient-Derived Xenografts Can Be Utilized to Test Response to Targeted Therapies
3. Discussion
4. Materials and Methods
4.1. Establishment of Patient-Derived Xenografts
4.1.1. Human Tissue Collection
4.1.2. Tissue Processing
4.1.3. Engraftment of Human Colorectal Cancer Tissue
4.1.4. Monitoring of Patient-Derived Xenograft Tumor Growth
4.1.5. Therapeutic Treatment of Patient-Derived Xenografts
4.2. Histological and Immunohistochemical Analysis
4.2.1. Tissue Fixation and Embedding
4.2.2. Histopathological Analysis of Patient-Derived Xenograft Lines
4.2.3. Immunohistochemistry
4.2.4. Imaging
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., III; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Hennessey, P.T.; Ochs, M.F.; Mydlarz, W.W.; Hsueh, W.; Cope, L.; Yu, W.; Califano, J.A. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Hogenesch, H.; Nikitin, A.Y. Challenges in pre-clinical testing of anti-cancer drugs in cell culture and in animal models. J. Control. Release 2012, 164, 183–186. [Google Scholar] [CrossRef] [Green Version]
- Houshdaran, S.; Hawley, S.; Palmer, C.; Campan, M.; Olsen, M.N.; Ventura, A.P.; Knudsen, B.S.; Drescher, C.W.; Urban, N.D.; Brown, P.O.; et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.J.; Beal, K.M.; DeGruttola, H.S.; Brennan, S.; Marzilli, L.A.; Anderson, K. Utilization of sequence variants as biomarkers to analyze population dynamics in cloned cell lines. Biotechnol. Bioeng. 2017, 114, 1744–1752. [Google Scholar] [CrossRef]
- Horbach, S.; Halffman, W. The ghosts of HeLa: How cell line misidentification contaminates the scientific literature. PLoS ONE 2017, 12, e0186281. [Google Scholar] [CrossRef]
- Otto, R.; Sers, C.; Leser, U. Robust in-silico identification of cancer cell lines based on next generation sequencing. Oncotarget 2017, 8, 34310–34320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, L.; Glanzel, W.; Korch, C.; Capes-Davis, A. Widespread Use of Misidentified Cell Line KB (HeLa): Incorrect Attribution and Its Impact Revealed through Mining the Scientific Literature. Cancer Res. 2017, 77, 2784–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, R.G.; Cobb, L.M.; Steel, G.G. The growth kinetics of xenografts of human colorectal tumours in immune deprived mice. Br. J. Cancer 1975, 31, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, J.A.; Taylor, D.M. Maintenance of biological and biochemical characteristics of human colorectal tumours during serial passage in immune-deprived mice. Br. J. Cancer 1978, 37, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiebig, H.H.; Schuchhardt, C.; Henss, H.; Fiedler, L.; Lohr, G.W. Comparison of tumor response in nude mice and in the patients. Behring Inst. Mitt. 1984, 343–352. [Google Scholar]
- Byrne, A.T.; Alferez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinska, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Du Manoir, S.; Orsetti, B.; Bras-Goncalves, R.; Nguyen, T.T.; Lasorsa, L.; Boissiere, F.; Massemin, B.; Colombo, P.E.; Bibeau, F.; Jacot, W.; et al. Breast tumor PDXs are genetically plastic and correspond to a subset of aggressive cancers prone to relapse. Mol. Oncol. 2014, 8, 431–443. [Google Scholar] [CrossRef]
- Goncalves, A.; Bertucci, F.; Guille, A.; Garnier, S.; Adelaide, J.; Carbuccia, N.; Cabaud, O.; Finetti, P.; Brunelle, S.; Piana, G.; et al. Targeted NGS, array-CGH, and patient-derived tumor xenografts for precision medicine in advanced breast cancer: A single-center prospective study. Oncotarget 2016, 7, 79414–79427. [Google Scholar] [CrossRef]
- Ni, J.; Ramkissoon, S.H.; Xie, S.Z.; Goel, S.; Stover, D.G.; Guo, H.B.; Luu, V.; Marco, E.; Ramkissoon, L.A.; Kang, Y.J.; et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat. Med. 2016, 22, 723–726. [Google Scholar] [CrossRef]
- Ionkina, A.A.; Tentler, J.J.; Kim, J.; Capasso, A.; Pitts, T.M.; Ryall, K.A.; Howison, R.R.; Kabos, P.; Sartorius, C.A.; Tan, A.C.; et al. Efficacy and Molecular Mechanisms of Differentiated Response to the Aurora and Angiogenic Kinase Inhibitor ENMD-2076 in Preclinical Models of p53-Mutated Triple-Negative Breast Cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Sinnwell, J.P.; Thompson, K.J.; Copland, J.A.; Marlow, L.A.; Miller, J.L.; Yin, P.; Gao, B.W.; et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Lee, H.C.; Seol, H.S.; Choi, Y.S.; Kim, E.; Lee, E.J.; Rhee, J.K.; Singh, S.R.; Jun, E.S.; Han, B.; et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget 2016, 7, 62533–62546. [Google Scholar] [CrossRef]
- Rios Perez, M.V.; Fleming, J.B. Patient-derived xenograft model of pancreatic cancer. In Patient Derived Tumor Xenograft Models: Promise, Potential and Practice; Academic Press: Cambridge, MA, USA, 2016; pp. 229–241. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of pancreatic cancer patient–derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin. Cancer Res. 2017, 23, 5639–5647. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, X.W.; Zhao, Z.Y.; Dong, B.; Guan, X.Y.; Tian, X.Y.; Qian, H.G.; Hao, C.Y. Establishment of pancreatic cancer patient-derived xenograft models and comparison of the differences among the generations. Am. J. Transl. Res. 2019, 11, 3128–3139. [Google Scholar] [PubMed]
- Moro, M.; Bertolini, G.; Caserini, R.; Borzi, C.; Boeri, M.; Fabbri, A.; Leone, G.; Gasparini, P.; Galeone, C.; Pelosi, G.; et al. Establishment of patient derived xenografts as functional testing of lung cancer aggressiveness. Sci. Rep. 2017, 7, 6689. [Google Scholar] [CrossRef]
- Drapkin, B.J.; George, J.; Christensen, C.L.; Mino-Kenudson, M.; Dries, R.; Sundaresan, T.; Phat, S.; Myers, D.T.; Zhong, J.; Igo, P.; et al. Genomic and functional fidelity of small cell lung cancer patient-derived xenografts. Cancer Discov. 2018, 8, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhao, J.; Zhang, Y.; Li, K.; Li, T.; Chen, X.; Zhao, S.; Zhao, S.; Liu, K.; Dong, Z. Establishment of lung cancer patient-derived xenograft models and primary cell lines for lung cancer study. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.N.; Choi, J.W.; Shim, H.S.; Kim, J.; Kim, D.J.; Lee, C.Y.; Hong, M.H.; Park, S.Y.; Park, A.Y.; Shin, E.J.; et al. Establishment of a platform of non-small-cell lung cancer patient-derived xenografts with clinical and genomic annotation. Lung Cancer 2018, 124, 168–178. [Google Scholar] [CrossRef]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef] [Green Version]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef]
- Heo, E.J.; Cho, Y.J.; Cho, W.C.; Hong, J.E.; Jeon, H.K.; Oh, D.Y.; Choi, Y.L.; Song, S.Y.; Choi, J.J.; Bae, D.S.; et al. Patient-derived xenograft models of epithelial ovarian cancer for preclinical studies. Cancer Res. Treat. 2017, 49, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of patient-derived tumor xenograft models of epithelial ovarian cancer for preclinical evaluation of novel therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, F.; Fratelli, M.; Guffanti, F.; Porcu, L.; Spriano, F.; Dell’Anna, T.; Fruscio, R.; Damia, G. Patient-derived ovarian cancer xenografts re-growing after a cisplatinum treatment are less responsive to a second drug re-challenge: A new experimental setting to study response to therapy. Oncotarget 2017, 8, 7441–7451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eoh, K.J.; Chung, Y.S.; Lee, S.H.; Park, S.A.; Kim, H.J.; Yang, W.; Lee, I.O.; Lee, J.Y.; Cho, H.; Chay, D.B.; et al. Comparison of clinical features and outcomes in epithelial ovarian cancer according to tumorigenicity in patient-derived xenograft models. Cancer Res. Treat. 2018, 50, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.; Chen, X.; Jiang, X.; Li, Z.; Yu, X.; Jin, K.; Cao, J.; Teng, L. Establishing a patient-derived colorectal cancer xenograft model for translational research. Int. J. Clin. Exp. Med. 2016, 9, 21346–21357. [Google Scholar]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Julien, S.; Merino-Trigo, A.; Lacroix, L.; Pocard, M.; Goeŕé, D.; Mariani, P.; Landron, S.; Bigot, L.; Nemati, F.; Dartigues, P.; et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. 2012, 18, 5314–5328. [Google Scholar] [CrossRef] [Green Version]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Puig, I.; Chicote, I.; Tenbaum, S.P.; Arqués, O.; Herance, J.R.; Gispert, J.D.; Jimenez, J.; Landolfi, S.; Caci, K.; Allende, H.; et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin. Cancer Res. 2013, 19, 6787–6801. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.P.; Calvo, E.; Ordonez, E.; Wick, M.J.; Viqueira, B.R.; Lopez-Casas, P.P.; Bruckheimer, E.; Calles-Blanco, A.; Sidransky, D.; Hidalgo, M. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, e45–e48. [Google Scholar] [CrossRef] [Green Version]
- Sargent, D.J.; Goldberg, R.M.; Jacobson, S.D.; Macdonald, J.S.; Labianca, R.; Haller, D.G.; Shepherd, L.E.; Seitz, J.F.; Francini, G. A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N. Engl. J. Med. 2001, 345, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, A.; Charette, M.L.; Maroun, J.; Brouwers, M.C.; Zuraw, L. Adjuvant therapy for stage II colon cancer: A systematic review from the Cancer Care Ontario Program in evidence-based care’s gastrointestinal cancer disease site group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Campbell, M.E.; Goldberg, R.M.; Grothey, A.; Seitz, J.F.; Benedetti, J.K.; Andre, T.; Haller, D.G.; Sargent, D.J. Survival following recurrence in stage II and III colon cancer: Findings from the ACCENT data set. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- Giacchetti, S.; Perpoint, B.; Zidani, R.; Le Bail, N.; Faggiuolo, R.; Focan, C.; Chollet, P.; Llory, J.F.; Letourneau, Y.; Coudert, B.; et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Vincenzi, B.; Imperatori, M.; Picardi, A.; Vespasiani, U.; Gallo, P.; Fausti, V.; Ceruso, M.S.; Santini, D.; Tonini, G. Liver toxicity in colorectal cancer patients treated with first-line FOLFIRI-containing regimen: A single institution experience. Expert Rev. Anticancer Ther. 2015, 15, 971–976. [Google Scholar] [CrossRef]
- Araghi, M.; Soerjomataram, I.; Jenkins, M.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Steele, S.R.; Park, G.E.; Johnson, E.K.; Martin, M.J.; Stojadinovic, A.; Maykel, J.A.; Causey, M.W. The impact of age on colorectal cancer incidence, treatment, and outcomes in an equal-access health care system. Dis. Colon Rectum 2014, 57, 303–310. [Google Scholar] [CrossRef]
- Moiel, D.; Thompson, J. Early detection of colon cancer-the kaiser permanente northwest 30-year history: How do we measure success? Is it the test, the number of tests, the stage, or the percentage of screen-detected patients? Perm. J. 2011, 15, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.C.; Mittal, P.K.; Sullivan, P.S.; Rutherford, R.; Staley, C.A.; Cardona, K.; Hawk, N.N.; Dixon, W.T.; Kitajima, H.D.; Kang, J.; et al. Colorectal Cancer Initial Diagnosis: Screening Colonoscopy, Diagnostic Colonoscopy, or Emergent Surgery, and Tumor Stage and Size at Initial Presentation. Clin. Colorectal Cancer 2016, 15, 67–73. [Google Scholar] [CrossRef]
- Chao, C.; Widen, S.G.; Wood, T.G.; Zatarain, J.R.; Johnson, P.; Gajjar, A.; Gomez, G.; Qiu, S.; Thompson, J.; Spratt, H.; et al. Patient-derived Xenografts from Colorectal Carcinoma: A Temporal and Hierarchical Study of Murine Stromal Cell Replacement. Anticancer Res. 2017, 37, 3405–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Samuels, Y. Molecular pathways: Dysregulated glutamatergic signaling pathways in cancer. Clin. Cancer Res. 2012, 18, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef]
- Bellone, G.; Smirne, C.; Carbone, A.; Buffolino, A.; Scirelli, T.; Prati, A.; Solerio, D.; Pirisi, M.; Valente, G.; Nano, M.; et al. KIT/stem cell factor expression in premalignant and malignant lesions of the colon mucosa in relationship to disease progression and outcomes. Int. J. Oncol. 2006, 29, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Sargent, D.; Sobrero, A.; Grothey, A.; O’Connell, M.J.; Buyse, M.; Andre, T.; Zheng, Y.; Green, E.; Labianca, R.; O’Callaghan, C.; et al. Evidence for cure by adjuvant therapy in colon cancer: Observations based on individual patient data from 20,898 patients on 18 randomized trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 872–877. [Google Scholar] [CrossRef]
- Ruiz de Garibay, G.; Mateo, F.; Stradella, A.; Valdes-Mas, R.; Palomero, L.; Serra-Musach, J.; Puente, D.A.; Diaz-Navarro, A.; Vargas-Parra, G.; Tornero, E.; et al. Tumor xenograft modeling identifies an association between TCF4 loss and breast cancer chemoresistance. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Li, J.; Wang, L.; Xu, J.; Cheng, Y.; Bai, Y.; Li, W.; Xu, N.; Lin, L.Z.; Wu, Q.; et al. Efficacy and Tolerability of First-Line Cetuximab Plus Leucovorin, Fluorouracil, and Oxaliplatin (FOLFOX-4) Versus FOLFOX-4 in Patients With RAS Wild-Type Metastatic Colorectal Cancer: The Open-Label, Randomized, Phase III TAILOR Trial. J. Clin. Oncol. 2018, 36, 3031–3039. [Google Scholar] [CrossRef]
- Ron, D.A.; Vera, R.; Labandeira, C.M.; Manrique, M.C.A.; Nunez, M.A.; Cid, N.G.; Mata, J.G.; Montes, A.F. Maintenance treatment in metastatic colorectal cancer: In search of the best strategy. Clin. Transl. Oncol. 2020, 22, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar]
- Collins, A.T.; Lang, S.H. A systematic review of the validity of patient derived xenograft (PDX) models: The implications for translational research and personalised medicine. PeerJ 2018, 6, e5981. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.; Vrignaud, P.; Vacher, S.; Richon, S.; Lièvre, A.; Cacheux, W.; Weiswald, L.B.; Massonnet, G.; Chateau-Joubert, S.; Nicolas, A.; et al. Evaluating patient-derived colorectal cancer xenografts as preclinical models by comparison with patient clinical data. Cancer Res. 2015, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Mackay, H.J.; Haluska, P. Patient-derived xenograft models in gynecologic malignancies. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e258–e266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Ahmed, S.; Adeyi, O.; Dick, J.E.; Ghanekar, A. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein-Barr virus. PLoS ONE 2012, 7, e39294. [Google Scholar] [CrossRef]
- Bondarenko, G.; Ugolkov, A.; Rohan, S.; Kulesza, P.; Dubrovskyi, O.; Gursel, D.; Mathews, J.; O’Halloran, T.V.; Wei, J.J.; Mazar, A.P. Patient-Derived Tumor Xenografts Are Susceptible to Formation of Human Lymphocytic Tumors. Neoplasia 2015, 17, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Butler, K.A.; Hou, X.N.; Becker, M.A.; Zanfagnin, V.; Enderica-Gonzalez, S.; Visscher, D.; Kalli, K.R.; Tienchaianada, P.; Haluska, P.; Weroha, S.J. Prevention of Human Lymphoproliferative Tumor Formation in Ovarian Cancer Patient-Derived Xenografts. Neoplasia 2017, 19, 628–636. [Google Scholar] [CrossRef]
- Fujii, E.; Kato, A.; Chen, Y.J.; Matsubara, K.; Ohnishi, Y.; Suzuki, M. Characterization of EBV-related lymphoproliferative lesions arising in donor lymphocytes of transplanted human tumor tissues in the NOG mouse. Exp. Anim. 2014, 63, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Khaled, W.T.; Liu, P. Cancer mouse models: Past, present and future. Semin. Cell Dev. Biol. 2014, 27, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosfjord, E.; Lucas, J.; Li, G.; Gerber, H.P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014, 91, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Uronis, J.M.; Osada, T.; McCall, S.; Yang, X.Y.; Mantyh, C.; Morse, M.A.; Lyerly, H.K.; Clary, B.M.; Hsu, D.S. Histological and molecular evaluation of patient-derived colorectal cancer explants. PLoS ONE 2012, 7, e38422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Maykel, J.; Liu, J.H.; Li, H.; Shultz, L.D.; Greiner, D.L.; Houghton, J. NOD-scidIl2rg tm1Wjl and NOD-Rag1 null Il2rg tm1Wjl: A model for stromal cell-tumor cell interaction for human colon cancer. Dig. Dis. Sci. 2014, 59, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Sanz, L.; Cuesta, A.M.; Salas, C.; Corbacho, C.; Bellas, C.; Alvarez-Vallina, L. Differential transplantability of human endothelial cells in colorectal cancer and renal cell carcinoma primary xenografts. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Fitzgibbon, M.P.; Mortales, C.L.L.; Towlerton, A.M.H.; Upton, M.P.; Yeung, R.S.; McIntosh, M.W.; Warren, E.H. Phenotypic and transcriptional fidelity of patient-Derived colon cancer xenografts in immune-deficient mice. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Hylander, B.L.; Punt, N.; Tang, H.; Hillman, J.; Vaughan, M.; Bshara, W.; Pitoniak, R.; Repasky, E.A. Origin of the vasculature supporting growth of primary patient tumor xenografts. J. Transl. Med. 2013, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Katsiampoura, A.; Raghav, K.; Jiang, Z.Q.; Menter, D.G.; Varkaris, A.; Morelli, M.P.; Manuel, S.; Wu, J.; Sorokin, A.V.; Rizi, B.S.; et al. Modeling of patient-derived xenografts in colorectal cancer. Mol. Cancer Ther. 2017, 16, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killion, J.J.; Radinsky, R.; Fidler, I.J. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998, 17, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chapman, W.; Hedley, D.W.; Oza, A.M.; Tannock, I.F. Assessment of tumor cell repopulation after chemotherapy for advanced ovarian cancer: Pilot study. Cytom. Part A J. Quant. Cell Sci. 2003, 51A, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gamarra-Luques, C.D.; Goyeneche, A.A.; Hapon, M.B.; Telleria, C.M. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Niida, A.; Uchi, R.; Hirata, H.; Komatsu, H.; Sakimura, S.; Hayashi, S.; Nambara, S.; Kuroda, Y.; Ito, S.; et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef]
- Lu, Y.W.; Zhang, H.F.; Liang, R.; Xie, Z.R.; Luo, H.Y.; Zeng, Y.J.; Xu, Y.; Wang, L.M.; Kong, X.Y.; Wang, K.H. Colorectal Cancer Genetic Heterogeneity Delineated by Multi-Region Sequencing. PLoS ONE 2016, 11, e0152673. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.M.; Donoho, G.P.; Iversen, P.W.; Zhang, Y.; Van Horn, R.D.; Forest, A.; Novosiadly, R.D.; Webster, Y.W.; Ebert, P.; Bray, S.; et al. Mouse PDX Trial Suggests Synergy of Concurrent Inhibition of RAF and EGFR in Colorectal Cancer with BRAF or KRAS Mutations. Clin. Cancer Res. 2017, 23, 5547–5560. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdirahman, S.M.; Christie, M.; Preaudet, A.; Burstroem, M.C.U.; Mouradov, D.; Lee, B.; Sieber, O.M.; Putoczki, T.L. A Biobank of Colorectal Cancer Patient-Derived Xenografts. Cancers 2020, 12, 2340. https://doi.org/10.3390/cancers12092340
Abdirahman SM, Christie M, Preaudet A, Burstroem MCU, Mouradov D, Lee B, Sieber OM, Putoczki TL. A Biobank of Colorectal Cancer Patient-Derived Xenografts. Cancers. 2020; 12(9):2340. https://doi.org/10.3390/cancers12092340
Chicago/Turabian StyleAbdirahman, Suad M., Michael Christie, Adele Preaudet, Marie C. U. Burstroem, Dmitri Mouradov, Belinda Lee, Oliver M. Sieber, and Tracy L. Putoczki. 2020. "A Biobank of Colorectal Cancer Patient-Derived Xenografts" Cancers 12, no. 9: 2340. https://doi.org/10.3390/cancers12092340
APA StyleAbdirahman, S. M., Christie, M., Preaudet, A., Burstroem, M. C. U., Mouradov, D., Lee, B., Sieber, O. M., & Putoczki, T. L. (2020). A Biobank of Colorectal Cancer Patient-Derived Xenografts. Cancers, 12(9), 2340. https://doi.org/10.3390/cancers12092340