1. Introduction
The P2X7 receptor (P2X7R) belongs to the family of ionotropic P2X receptors, which function as ATP-gated cation selective channels that are permeable to Na
+, Ca
2+ and K
+ ions. However, P2X7R displays several functional and biological specificities differing largely from the other members of the P2X receptor family [
1]. From a pharmacological point of view, a specific feature of P2X7R is to be 10–100 times less sensitive to its natural agonist ATP than all other P2X receptors, requiring millimolar concentrations to show measurable activation while all the others are activated by micromolar concentrations. In addition, P2X7R is about 10–30 times more sensitive to 2,3-O-(4-benzoylbenzoyl)-ATP (BzATP), a synthetic ATP analogue, than ATP [
1]. A unique functional property of P2X7R is that upon prolonged or repeated agonist stimulation, it exhibits no desensitisation and instead striking facilitation or sensitization, a functional property characterized by an increase in the current response as well as an enhanced agonist sensitivity [
2]. This unique functional property results in potentiation of downstream signalling pathways initiated by P2X7R.
While all other P2X receptors are mainly expressed in the nervous system, such as in neurons and glial cells, participating in the modulation of synaptic transmission, the P2X7R is mostly characterized by its predominant expression in immune cells and its critical role in immunity and inflammatory responses [
3]. In the context of cancers, the P2X7R has attracted escalating attention over the past years. It has been reported that P2X7R is overexpressed in many different types of tumours, specifically in miscellaneous carcinomas [
4], with the possible exception of cervical and endometrial cancers, in which it was intriguingly down-regulated [
5,
6]. Many studies provide evidence to show that P2X7R activity or activation protects cells from apoptosis [
7], promotes cancer cell trophic activity and primary tumour growth [
8,
9] and stimulates cancer cell migration or invasion [
10,
11,
12,
13,
14,
15]. These studies thus favour a pro-cancerous role for P2X7R and propose that P2X7R antagonists are promising anticancer drugs [
16]. In contrast, several other studies demonstrate that P2X7R induces cancer cell death upon ATP stimulation [
17] or even enhances anti-tumour immune responses from the host organism, thus supporting the P2X7R to be anti-cancerous [
18,
19]. The role of P2X7R in cancer promotion appear to depend on multiple factors, including the type of cancers or tumours, the level of expression or activation of the receptor, the cell type considered (cancer cells vs. immune cells), the level of immune cell infiltration within the tumour and also the phase in the carcinogenic progression. Therefore, it remains unclear whether it would be beneficial, or detrimental, to antagonize P2X7R for anticancer purposes, and this needs to be addressed in specific cancer models.
In breast cancer, it has been shown that the P2X7R is overexpressed [
20], fully functional and promotes cancer cell invasiveness, both in vitro and in an in vivo zebrafish model [
12,
21,
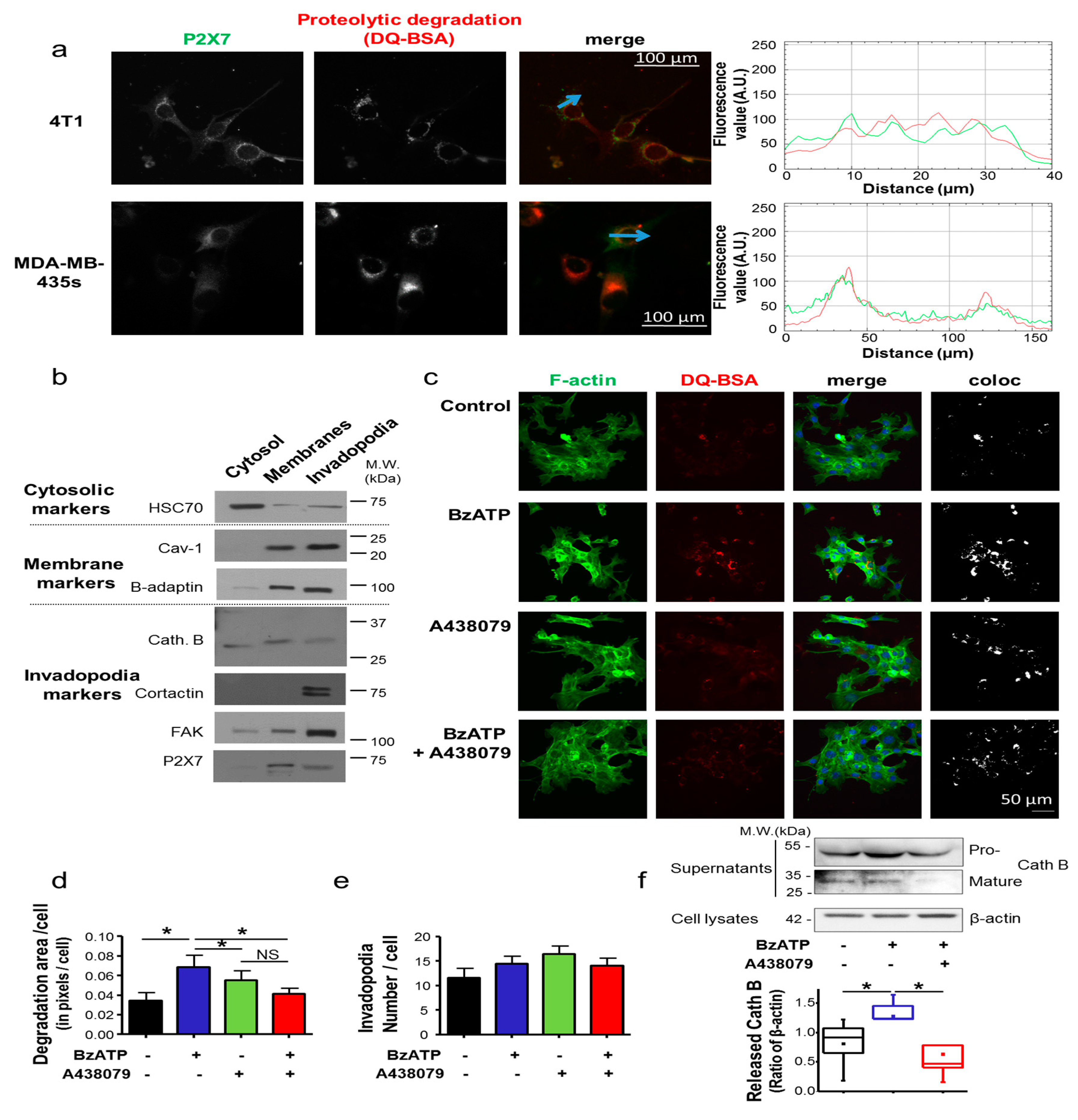
22]. Moreover, P2X7R was demonstrated to induce the release of active proteolytic cysteine cathepsins [
12], a finding that further suggests the involvement of P2X7R in the “mesenchymal mode” of invasion, in which cells remodel the extracellular matrix (ECM), by forming particular cellular structures that are enriched in F-actin and protrusive into the ECM called “invadopodia”, and using such proteolytic activity, generate their pathway [
23].
In this study, we assessed the role of P2X7R in mammary cancer cells in determining the invasive properties, using both 2- and 3-dimensions models, more specifically, the invadopodial activity, and characterized the involvement of P2X7R in the acquisition of a mesenchymal phenotype in vitro. We also assessed the consequences of P2X7R expression in primary tumour growth and metastatic spreading in vivo. This study further aimed to clarify the relevance of pharmacological intervention using specific P2X7R antagonists in the treatment of mammary cancers. For this purpose, we developed an orthotopic, syngeneic and immunocompetent mammary cancer model in BALB/cJ mice, and studied the role of P2X7 receptor, expressed or not in 4T1 mammary cancer cells (using genetically engineered cells) or in the host organisms (wild-type P2rx7+/+ versus knock-down P2rx7−/− mice). Our results unequivocally demonstrate that P2X7R is functionally expressed in mammary cancer cells and its activation promotes the acquisition of a mesenchymal phenotype and enhances invadopodial activity. Furthermore, we provide compelling evidence to indicate that the P2X7R expressed in mammary cancer cells but not in the host organism plays a key role in primary tumour growth and metastatic development, which are significantly attenuated by treatment with specific P2X7R antagonists. These findings support that the P2X7R in mammary cancer cells drives mammary tumour progression and represents a pertinent target for mammary cancer treatment.
3. Discussion
The acquisition of invasive capacities by cancer cells represents a critical mechanism in the cancer progression and metastasis that significantly impact the survival of cancer patients. Currently, there is no specific treatment for preventing cancer cell spreading from the primary tumour or preventing metastases appearance. At the cellular level, several modes of cancer cell migration and invasiveness have been described, both in in vitro and in vivo models. Among these are the “amoeboid” and the “mesenchymal” modes, that could be identified in both collective and individual cell invasions [
29]. Even though these modes of invasion could be more characteristic of some cancer types or subtypes, cancer cells do not necessarily stay or are engaged in a specific one. The most aggressive cells often have the ability to switch from one mode to the other, depending on biological, physical and chemical conditions and constrains of the microenvironments [
30]. Whatever the mode of invasion, being individual or collective, the capacity of degrading extracellular matrices that determines the dissemination rate of cancer cells critically depends on the activities of proteases, such as matrix metalloproteinases (MMP) and cathepsins. This capacity to invade extracellular matrices with a proteolytic degradation was initially described as being a feature of mesenchymal cancer cells. Mesenchymal cancer cells display an elongated fibroblast-like morphology, with a rear-to-front lamellopodial cell polarity, and harbour multiple cell-matrix adhesions, such as filopodial structures. The degradation of the ECM is recognized to be performed by invadosomal structures, which are F-actin-rich and protrusive into the ECM and responsible for its proteolysis through recruiting both membrane-associated and extracellularly-released soluble proteases [
23,
27].
Extracellular ATP is known to be important in the regulation of differentiation and activation of multiple cell types, under physiological conditions and also under pathological conditions. Over the past few years, ATP-induced purinergic signalling has attracted considerable attention in the field of oncology [
4,
31]. Indeed, ATP and other nucleotides have been demonstrated to be present in high concentrations in the tumour microenvironments, owing to the active release of ATP from cancer cells and to cell necrosis in the hypoxic halo of solid tumours [
32,
33]. When released in the extracellular compartment, ATP activates plasma membrane purinergic P2 receptors and downstream signalling pathways, which are well documented in different cell types in the tumour, such as cancer cells of course and also immune cells, fibroblasts and endothelial cells. Obviously, the consequences on tumour and disease progression could be drastically opposite, depending on the cell type considered, and therefore it is indispensable to develop a clear understanding of the diseases as a whole. The P2X7R is a very intriguing receptor, which has been demonstrated to be upregulated in multiple cancer types [
4], but its clear involvement in carcinogenesis or cancer progression is still debated. Because the over-stimulation of P2X7R, either in dose or in duration, is known to induce cell death [
34], several studies postulated that it might be non-functional in proliferating cancer cells [
20,
35]. It was even proposed that stimulating its activity in cancer cells would induce their death and could represent therapies for treating cancers [
36]. However, multiple studies demonstrated that the P2X7R is expressed and functional in cancer cells and that its basal activity or its external stimulation with biologically relevant concentrations of ATP support cancer cell growth, migration and invasion [
12,
13,
37], and tumour growth [
9,
14]. In these models, pharmacological antagonism of P2X7R appeared to be effective in reducing cancer progression, suggesting that targeting P2X7R could represents a novel opportunity for anticancer treatments.
It is now clearly established that the ATP concentration can reach mM in the necrotic areas of solid tumours. Such massive necrosis, which provides the large amount of ATP required to activate the P2X7R, mainly occurs in the inner core of the tumour (e.g., being ischemic and hypoxic) with a decreasing gradient toward the outer part of the tumour. The outer part is involved in local invasion and subsequent metastasis. Under such conditions, it is likely that excessive concentration of ATP in the necrotic core induces cancer cell death, while a moderate concentration of ATP present at the edge of the growing tumours stimulates invasion, through activating the P2X7R in cancer cells, as proposed in the “run or die” hypothesis [
16]. Also, it is not well understood but it may be important for further studies to assess the P2X7R expression within the tumour and compare the expression level in the inner core and the outer layer of the tumour.
On the other hand, the important role of P2X7R in the immune responses is well established [
3]. Its expression and activity in cells from the host organism of a tumour might interfere with tumour progression. Importantly, its role in tumour-associated dendritic cells for the presentation of tumour antigen to CD4
+ lymphocytes, and its role in anti-tumour immune responses were demonstrated [
18,
38]. Therefore, it was questioned whether P2X7R had a real role in cancer progression, as well as whether using P2X7 antagonists was pertinent and useful for anticancer treatment. In a very nice study, Adinolfi and collaborators [
39] demonstrated that, when expressed in mouse melanoma or colon cancer cells subcutaneously inoculated to P2X7R-deficient mice, P2X7R strongly promoted tumour growth and metastatic dissemination. However, both primary tumour growth and metastases were reduced in P2X7R-expressing wild-type mice because of a P2X7R-dependent anti-tumour immune response [
39]. Another study, performed in the context of colon cancer, also supports an important role for P2X7R in anti-tumour immune response. In this study, Hofman and collaborators [
40] showed that pharmacological antagonism or genetic silencing of the P2X7R altered immune cell infiltration and increased tumour incidence in a mouse model of colitis. Therefore, from these studies it appeared that the use of P2X7 antagonists for anti-cancer treatment might be deleterious and could lead to opposite tumour-promoting effects. Melanoma and colon cancer development and progression are known to be highly dependent on the immune and inflammatory system [
41,
42]. For other cancers such as mammary cancer, the question is still open, and the relative participation of P2X7R expressed by cancer cells or host cells might be different and should be specifically assessed. Breast cancer is the primary cause of death by cancer in women worldwide and patients mostly die because of metastases appearance and development [
43]. Breast tumours are also highly heterogeneous and there are four major molecular subtypes differing for their prognostic and treatments: Luminal A, Luminal B, HER2-enriched and Triple negative/basal-like. Triple negative/basal-like tumours are often very aggressive and have a poorer prognosis compared to the ER-positive subtypes (luminal A and luminal B tumours) [
44].
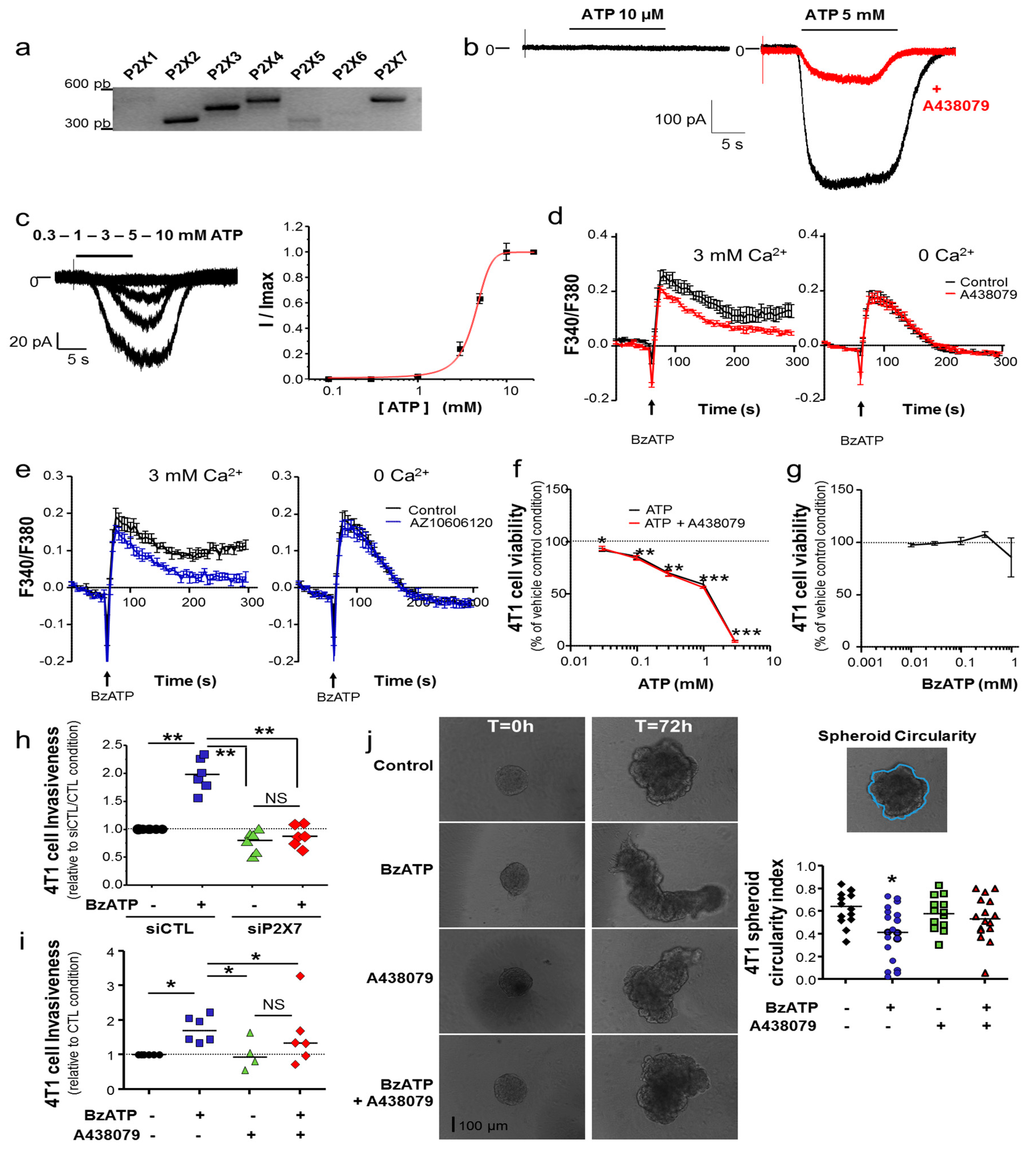
In this study, we have examined 4T1 mouse mammary cancer cells, which are a model of triple-negative mammary tumours. We demonstrated that the P2X7R is functional in 4T1 cancer cells and that its activation with natural (ATP) or synthetic (BzATP) agonists induces typical facilitating inward currents and increases in intracellular Ca
2+ levels. Its stimulation did not seem to induce cell death, even though the genetic silencing of the
P2rx7 gene led to a slight increase in the proliferation rate, and instead importantly enhanced cell invasiveness under 2D and 3D conditions. These effects were similarly prevented both by treatment with pharmacological antagonists and by genetic depletion, indicating that not only the P2X7R expression but also its activity is important in promoting cell invasiveness. These results are in agreement with previous reports performed with human cancer cells [
11,
12,
13,
15,
22,
45]. While mechanistic determinants for such a pro-invasive role were not clearly delineated, we have identified for the first time in this study the P2X7R expression in invadopodial structures involved in ECM degradation. More specifically, the P2X7R expression enhanced the ECM-degradative activity of invadopodia, most likely through stimulating release of proteolytic enzymes [
12] such as Cath B (
Figure 2f), rather than formation of these structures
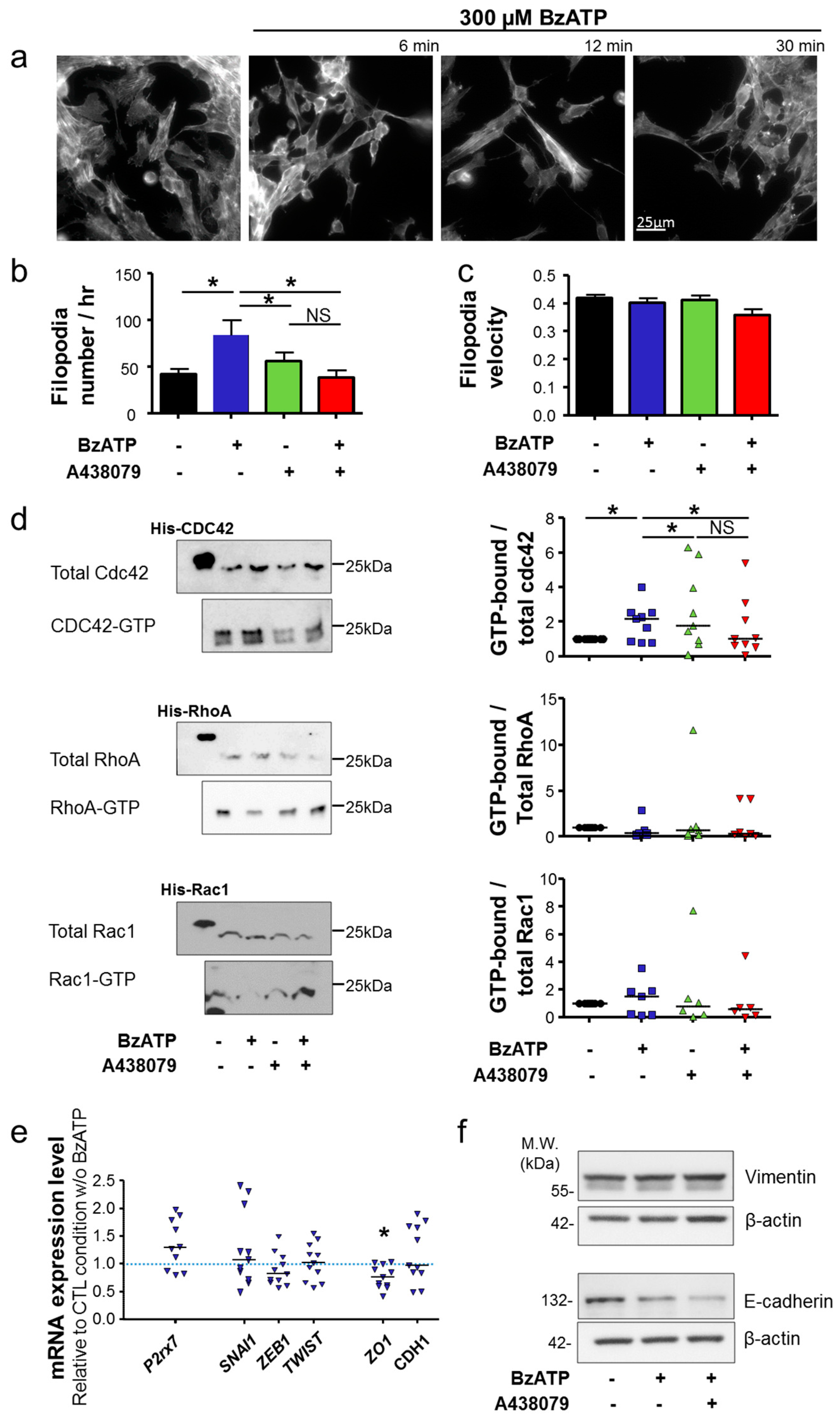
per se. Activation of the P2X7R also importantly modified cancer cell morphology and triggered the acquisition of a more aggressive phenotype, characterized by activation of Cdc42 Rho-GTPase, remodelling of F-actin, elongation of cells and formation of filopodia. Taken together, the results presented in this study strongly suggest the involvement of P2X7R in promoting the acquisition of a mesenchymal invasive phenotype in mammary cancer cells. Activation of P2X7R led to a reduction in the expression of
ZO-1, which is important in maintaining tight junction function and epithelial polarity. However, in our model, activation of the P2X7R appears not important in inducing EMT. Indeed, there was no significant change in the expression of EMT-promoting transcription factors, Zeb1, Snail1 or Twist, and the expression of the gene encoding for E-cadherin was not modified. The protein expression of the mesenchymal marker vimentin was not modified either. These findings are different from what are reported in recent studies examining other cancer types. In prostate cancer cells, P2X7R was shown to promote invasiveness and metastatic properties and silencing its expression attenuated ATP- or BzATP-induced changes in the expression of EMT-related genes, Snail, E-cadherin and Claudin-1 [
15]. In osteosarcoma cells, stimulation of P2X7R reduced the expression of E-cadherin, and induced the expression of Snail, vimentin and fibronectin. These effects were prevented by pharmacologically antagonizing the P2X7R or knocking down its expression [
45]. However, these differences might depend on the cell types studied and on their level of transition. Indeed, it was reported that murine 4T1 mammary cancer cells, which are highly invasive and metastatic, do not strictly exhibit the genotypic and phenotypic properties of EMT [
46]. It was postulated that other processes may govern the metastatic capability of these cells. Furthermore, it is know that EMT is not an all-or-nothing phenomenon, and some cancer cells display intermediate or partial transition states that have been identified to be highly aggressive [
47].
Here, we investigated the role of P2X7R, whether it was expressed by cancer cells or by host cells, in mammary cancer progression. By using
P2rx7+/
+ and
P2rx7−/
− mice, we demonstrated that, in the syngeneic and orthotopic model of mammary cancer, there is no participation of the P2X7R in the host organism in either anti- or pro-tumour activities. Because the level of the anti-tumour response of the host organism might depend on the size of the primary tumour, we inoculated two different numbers of mammary cancer cells (low density of 1 × 10
4 or high density of 1 × 10
6 cells per inoculation site) but no difference was observed. Such a finding is in apparent contradiction to what were reported by previous studies [
39,
40], but may be due to the specific mammary gland microenvironment [
48], as compared to the subcutaneous or colic environments, or to specific anti-immune responses induced by 4T1 cells. Such possibilities should be investigated in further studies. However, in stark contrast, the expression and activity of P2X7R in mammary cancer cells had a predominant effect in mammary tumour growth and metastasis development. Loss of the
P2rx7 expression in two cell lines derived from 4T1 cells led to a significant delay in primary tumour growth, while the cells demonstrated a slight increase in the proliferation rate compared to the
P2rx7+/+ cells. The consistent results using two different
P2rx7-knocked-out clones rule out possible off-targets of the CRISPR/Cas9 technique used and demonstrate the critical involvement of cancer cell invasiveness in primary tumour growth. It is interesting to notice that while knocking out the expression of P2X7R in 4T1 cells slightly increased their proliferation rate in vitro, it completely abolished the growth of the primary mammary tumour in vivo. One possible explanation is that, by promoting the proteolytic degradation of the ECM and local invasion, P2X7R importantly supports primary tumour growth. We also demonstrated a clear reduction of metastatic colonization of organs when cancer cells did not express the
P2rx7 gene. However, it remains unclear whether this effect is specific or due to the important delay in primary tumour growth. Furthermore, we demonstrated that treatment with specific P2X7 antagonists with different modes of action, one being competitive (A438079) and the other being non-competitive (AZ10606120), had similar effects in reducing the tumour growth in wild-type mice.
4. Materials and Methods
4.1. Agonists, Antagonists, Salts and Chemicals
Adenosine 5′-triphosphate (ATP) disodium salt and 2′-(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate (BzATP) triethylammonium salt were purchased from Sigma-Aldrich (St. Quentin Fallavier, France) and prepared in PBS without Ca2+. A438079, AZ10606120 and NF340 were purchased from Tocris Bio-Techne (Noyal Châtillon sur Seiche, France) and prepared in saline solution. All salts and pH buffers for electrophysiological solutions were purchased from Sigma-Aldrich. Fluorescent probes (Phalloidin DyLight® 488, DQ™-BSA and Fura2-AM) were all purchased from Invitrogen (Thermo Fisher Scientific, Villebon-sur-Yvette, France).
4.2. Cells and Cell Culture
Human melanoma cancer cell line MDA-MB-435s-luc (thereafter called “MDA-MB-435s”) was constructed as previously described [
49]. Murine mammary cancer cell line 4T1 from the Balb/c strain was purchased from LGC Standards (Molsheim, France), and a stable 4T1-luc cell line expressing the luciferase gene (thereafter called “4T1 cells”) was obtained by transduction with lentiviral vectors containing the luciferase gene and blasticidin resistance gene for selection (GIGA Viral Vectors, Liege, Belgium). stable 4T1 cell lines knocked-out for the expression of the
P2rx7 gene were obtained using the CRISPR/Cas9 technique by transfection with the
P2rx7 Double Nickase Plasmid (Santa Cruz Biotechnology, CliniSciences, Nanterre, France). Clonal selection was performed using 2 µg/mL puromycin. Two clones have been kept for this study, called “Crispr#1” and “Crispr#2”. A null-target Double Nickase Plasmid was also used to transfect 4T1 cells, leading to the selection of a control cell line, thereafter called “CTL” cell line. Efficiency of the CRISPR-mediated knock-down was assessed by RT-qPCR and invasion assays, and stability of clones was followed for a minimal duration of 6 weeks. Selected clones displayed similar levels of luciferase activity.
MDA-MB-435s cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% foetal calf serum (FCS). All 4T1-derived cells were cultured in RPMI medium supplemented with 10% FCS. Cells were grown at 37 °C in a humidified 5% CO2 incubator. Mycoplasma contamination tests were performed routinely (MycoAlert™ Mycoplasma Detection Kit, Lonza, Thermo Fisher Scientific, Villebon-sur-Yvette, France).
4.3. Small Interfering RNA Transfection
4T1 mammary cancer cells were transfected with siRNA directed against mouse P2rx7 mRNA (siP2X7) or scrambled siRNA as a control (siCTL), both of which were purchased from Tebu-Bio (Le Perray-en-Yvelines, France). Cells were transfected with 20 nM siRNA by using Lipofectamine RNAi max (Invitrogen). Experiments were performed 24 h after transfection and efficacy of silencing was assessed by RT-qPCR.
4.4. RNA Extraction, Reverse Transcription and Polymerase Chain Reaction
For conventional RT-PCR, total RNA was extracted using NucleoSpin® RNA II kit (Macherey Nagel EURL, Hoerdt, France) and reverse transcribed with PrimeScript™ RT Reagent (Ozyme, Saint-Cyr-l’École, France). PCR was performed with GoTaq® Flexi DNA Polymerase (Promega, Charbonnières-les-Bains, France) according to manufacturer’s recommendations. PCR products were loaded on 2% agarose gels and visualised after UV excitation.
For quantitative PCR (qPCR), total RNA was extracted using TRIzol™ Reagent (Invitrogen), and reverse-transcribed with the PrimeScript™ RT Reagent Kit (Ozyme, France). PCR were performed using SYBR qPCR Premix Ex Taq (Ozyme, France) and CFX CONNECT (Bio-rad, Marnes-la-Coquette, France). All primers sequences are described in
Table 1.
4.5. Electrophysiology
Electrophysiological recordings of ATP-induced ionic currents were performed in the whole- cell configuration of the patch clamp technique. Patch pipettes were pulled from borosilicate glass (World Precision Instruments, Hitchin, UK) to a resistance of 4–6 MΩ. Currents were recorded under voltage-clamp mode using an Axopatch 200B amplifier (Axon Instrument, Molecular Devices, San Jose, CA, USA) and analogical signals were filtered at 10 kHz and digitized using a 1322A Digidata converter (Molecular Devices, San Jose, CA, USA). Cell capacitance and series resistance were electronically compensated. Membrane potential was held at −60 mV. Experiments were performed at room temperature (20–25 °C) in extracellular physiological saline solution (PSS in mM: 147 NaCl, 10 N-2-hydroxyethylpiperazine-N′-2ethansulphonic acid (HEPES), 13 D-glucose, 2 KCl, 2 CaCl
2 and 1 MgCl
2) and pipettes were filled with intracellular saline solution (in mM: 147 NaCl, 10 HEPES and 10 ethylene glycol-bis-(2-aminoethyl ether)-N, N, N′, N′-tetraacetic acid (EGTA) with osmolarity and pH values of 295–315 mOsm and 7.3, respectively). ATP was externally applied using a RSC160 fast-flow delivery system (BioLogic Science Instruments, Seyssinet-Pariset, France) for 10 s, at the concentrations indicated in the
Figure 1 legend. P2X7 antagonist A438079 was perfused into the bath for 2 min before recording its effect in the presence of 5 mM ATP. ATP concentration-response curves were obtained by first obtaining a maximum response to 10 mM and then by applying decreasing concentrations of ATP and the results were plotted using Origin Pro 2015 software (Microcal Software Inc., Northampton, MA, USA). EC
50 was derived by fitting the data to Hill equation provided by the software.
4.6. Intracellular Ca2+ Measurement
ATP- and BzATP-induced increases in intracellular Ca
2+ levels were measured using ratiometric fluorescent probe Fura2-AM and a Flexstation3 microplate reader (Molecular Devices) as previously described [
25]. Cells were incubated with 1 µM Fura2-AM for 45 min in OptiMEM medium at 37 °C prior to conduct recordings. Cells were washed and then pre-incubated in the extracellular saline solution (in mM: 147 NaCl, 2 KCl, 1 MgCl
2, 10 HEPES and 13 D-glucose, pH 7.4) with and without 3 mM CaCl
2. F340/F380 defined the ratio of the fluorescence intensity excited alternatively at 340 nm and 380 nm and emitted at 510 nm and was used to indicate the free intracellular Ca
2+ concentration. 3 mM ATP or 0.3 mM BzATP was applied to cells after the baseline was established. Antagonists, A438079 (10 µM) or AZ10606120 (300 nM) were added 5 min before application of agonist.
4.7. Cell Viability
The effects of exposure to ATP and BzATP at doses indicated in the figure legends for 24 h on 4T1 cell viability were evaluated by using the tetrazolium salt assay (MTT) as previously described [
12]. Briefly, cells were treated with increasing doses of ATP and BzATP for 24 h and cell viability was measured after incubation with MTT for 40 min at 37 °C.
4.8. Cell Adhesion
To assess 4T1 cancer cell adhesion, 2 × 104 cells were seeded in their normal culture medium in wells of a 96-well plate and placed in the incubator for 30 min at 37 °C and 5% CO2. After one wash in PBS, the number of adherent cells was evaluated in wells using the MTT assay.
4.9. Two- and Three-Dimensions In Vitro Invasion Assays
2-D cancer cell invasiveness was measured as previously described [
12] using 8 µm pore-size polyethylene terephtalate membrane inserts covered with Matrigel™ matrix (Becton Dickinson, Le Pont de Claix, France). Cells at the lower surface of the insert were stained with DAPI and nuclei were counted after collecting pictures with a Nikon TI-S microscope (Nikon S.A.S., Champigny-sur-Marne, France). Results were normalized to the control condition in the absence of agonist or antagonist.
For 3D invasiveness assays, cancer cell spheroids were used. To do so, 500 4T1 cells or 2000 MDA-MB-435s cells were seeded in wells of ultra-low attachment 96-well plates (Corning, Boulogne-Billancourt, France). After allowing spheroid formation at 37 °C and 5% CO2 in the incubator for 24 h for 4T1 and 48 h for MDA-MB-435s, 4 mg/mL Matrigel™ was added to the wells, in the absence or presence of 0.3 mM BzATP, and 10 µM A438079. 3D-Matrigel invasion by spheroids was followed every hour using a Nikon TI-S microscope. Invasion distance and spheroid circularity were measured and calculated using ImageJ version 1.48.
4.10. Invadopodia Activity Assay
Invadopodia activity was assessed by culturing cells for 24 h on the top of a layer of Matrigel™ (4 mg/mL) matrix containing 50 µg/mL of DQ-BSA in LabTeck™ (Palaiseau, France) chambers, as previously reported [
26]. Briefly, cells were fixed in 4% paraformaldehyde for 15 min, permeabilized using 0.02% saponin for 20 min and incubated with 3% bovine serum albumin (BSA) for 30 min. F-actin was stained with 1.5 units/mL Phalloidin DyLight 488 (Invitrogen). Slides were mounted using ProLong
® Gold Antifade Mountant with DAPI (Invitrogen). Epifluorescence microscopy was performed with a Nikon TI-S (Nikon). Co-localized pixels for DQ-BSA and Phalloidin-488 were obtained using NIS-Element (Nikon) and fluorescence intensity, as well as the number of invadopodia, was quantified with ImageJ.
4.11. Invadopodia Fractionation
Invadopodia, which were embedded in the gelatin matrix, were separated from cellular bodies using the previously described protocols [
26]. Briefly, cells were grown on the top of a 2%-gelatin matrix. Cellular bodies were removed using osmotic shock and further fractionated to isolate a “cytosolic” and an “all membranes” fractions. Proteins from invadopodia were solubilised in a lysis buffer containing 0.1% NP-40 and 1 mM DTT and separated from gelatin by centrifugation at 17,000×
g for 30 min. Proteins in each fraction were separated according to standard SDS-PAGE protocols on 8% and 12% polyacrylamide gels and then transferred on PVDF membrane. Primary antibodies used were: mouse anti-HSC70 (sc-7298 Santa Cruz Biotechnology, Inc., Heidelberg, Germany), rabbit anti-caveolin 1 (sc-894 Santa Cruz Biotechnology, Inc., Germany), rabbit anti-β-adaptin (610382 BD Biosciences, Le Pont de Claix, France), cathepsin B (20-CR71 Fitzgerald, Acton, MA, USA), cortactin (05-180 Millipore, Guyancourt, France), FAK (036SC-558 Santa Cruz Biotechnology, Inc., Heidelberg, Germany), P2X7 (APR-008 Alomone Labs Ltd., Jerusalem, Israel). Secondary HRP-conjugated antibodies were: goat anti-mouse (Santa Cruz Biotechnology, Inc., Heidelberg, Germany), goat anti-rabbit (Jackson Immunoresearch Interchim, Montluçon, France), rabbit anti-β-actin-HRP (Santa Cruz Biotechnology, Inc., Heidelberg, Germany). Densitometric analyses were performed using ImageJ. Full uncropped blots are shown in
Figures S5 and S6.
4.12. Epifluorescence Experiments
Cells were grown for 24 h in LabTeck™ chambers on a layer of Matrigel matrix (4 mg/mL) containing 50 µg/mL of DQ-BSA. Cells were fixed in 4% paraformaldehyde for 15 min and then incubated with 3% BSA for 30 min. P2X7 was immunodetected using a primary antibody (APR-008, Alomone Labs Ltd., Israel) and a secondary anti-rabbit IgG AlexaFluor488 (Invitrogen, France). For actin cytoskeleton analysis, cells were grown on glass coverslips until 40% confluency and then serum was reduced to 1% for 24 h and 0% for 24 h. Cells were treated with BzATP for 2, 4, 6, 12, 30 min and 24 h. Cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.1% triton-X-100 for 5 min, incubated with 3% BSA for 30 min and stained with 1.5 units/mL Phalloidin DyLight® 488 (Invitrogen, France) for 1 h. Slides were mounted using ProLong® Gold Antifade Mountant with DAPI (Invitrogen, France). For the analysis of filopodia, cells were transfected with Ibidi® LifeAct plasmid (Generously provided by Laurent Counillon, CNRS UMR7370, University of Nice-Sophia Antipolis, Nice, France) using TransIT®-2020 (Mirus Euromedex, Souffelweyersheim, France). Epifluorescence and time-lapse microscopy was performed with a Nikon TI-S (France) (see Videos S1–4). Numbers of filopodia formed per hour and filopodia velocity were quantified using the Adapt plugin for ImageJ.
4.13. RhoGTPases Pull-Down Assays
The activity of RhoA, Rac1 and Cdc42 was assessed using pull-down assays according to the manufacturer’s protocols (Cat BK030 RhoA/ Rac1/Cdc42 Activation Assay Combo Biochem Kit, Cytoskeleton, Tebu-Bio, France) as previously described [
50]. Briefly, cells were grown until 40% confluency in 75 cm
2 flasks in normal growing medium, and serum was reduced to 1% for 24 h and 0% for 24 h. After treatment with 0.3 mM BzATP with or without 10 µM A438079 for 30 min in culture medium without serum, cells were lysed and samples were snap-frozen in liquid nitrogen. For each condition, 500 µg total lysates were used for pull-down assay according to the manufacturer’s instructions and then used for western blotting (Full uncropped blots are shown in
Figure S7).
4.14. Western-Blotting Experiments
Cells were washed twice with PBS and lysed in presence of a lysis buffer (50 mM Tris, pH 7, 100 mM NaCl, 5 mM MgCl2, 10% glycerol, 1 mM EDTA), containing 1% Triton-X-100 and protease inhibitors (Sigma-Aldrich, France). Cell lysates were cleared by centrifugation at 16,000×
g for 10 min. Total protein concentrations were determined using the Pierce
® BCA Protein Assay Kit (Thermofisher Scientific, Villebon-sur-Yvette, France). Protein sample buffer was added and samples were boiled at 100 °C for 3 min. Samples were loaded (20 μg of total proteins for cell lysates and 6 μL of concentrated supernatants) and electrophoretically separated on 10% or 12% polyacrylamide gels and then transferred to polyvinylidene difluoride membranes. Vimentin was detected using a primary anti-vimentin rabbit antibody (1/1000, clone D21H3, Cell Signaling Technology, Danvers, MA, USA) and a secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody at 1:2000 (TEBU-BIO, Le Perray-en-Yvelines, France). E-cadherin was detected using a monoclonal mouse primary antibody (1/1000, HECD-1, 13-1700 ThermoFisher Scientific) and a secondary HRP-conjugated anti-mouse-IgG secondary antibody at 1:2000 (TEBU-BIO). β-actin was used as a sample loading control using anti-β-actin-HRP primary antibody at 1:1000 (C4, Santa Cruz Biotechnology ref sc-47778). Western blot experiments for the detection of both pro and mature forms of cathepsin B were performed from 4T1 concentrated supernatants. Cells were grown to 80% confluence in a 6-well plate. After 24 h treatment (BzATP ± A438079) in Optimem medium (Invitrogen, France), cell supernatants were taken and concentrated using 10,000 MWCO filters (Merck Millipore, Molsheim, France). Cathepsin B proteins were visualized using polyclonal rabbit anti-human cathepsin B primary antibodies (1/150, Fitzgerald) and a goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1/5000).Proteins were revealed using electrochemiluminescence-plus kit (Pierce ECL Western Blotting Substrate, ThermoFisher Scientific, France) and captured on a PXi acquisitions system (SYNGENE, Cambridge, UK). Densitometry analysis of protein bands was performed using the Gel Tool from Fiji software (Scientific image analysis software available at
https://fiji.sc). Full uncropped blots are shown in
Figures S5 and S6.
4.15. In Vivo Mammary Cancer Model and Experiments
All experiments have been approved by the Comité d’éthique du Centre-Val de Loire (Comité d’éthique en expérimentation animale Campus CNRS d’Orléans n°3, Ref 005377.01 Apafis #12960) and were performed in accordance with the European Ethics rules. All animals were bred and housed in isolated ventilated cages at the CNRS UPS44—TAAM-CIPA (CNRS Campus, Orléans, France), in controlled conditions with a 12 h light/dark cycle at 22 °C, and food and water ad libitum. We developed a syngeneic and orthotopic mouse mammary cancer model in female BALB/cJ immunocompetent mice. To do so, 4T1-luciferase-expressing mouse mammary cancer cells were injected into the fifth mammary fat pad of 6 weeks-old mice. The luciferase activity was mainly used to follow secondary tumour appearance and growth in vivo, subsequent to D-luciferin (150 mg/kg) intraperitoneal injection and bioluminescent imaging (IVIS Lumina II, Perkin Elmer, Villebon-sur-Yvette, France). Primary tumour volume (mm3) and growth over time were most effectively measured with a calliper, twice a week, and calculated as (L × l2)/2 (in mm). Metastases were counted macroscopically at the completion of studies, during autopsies. Animal weight was measured once a week.
In experiments comparing the role of the number of injected cells on primary tumour growth, in mice expressing the
P2rx7 gene or not, cell suspensions of either 1 × 10
6 or 1 × 10
4 4T1-luc derived CTL (
P2rx7+/+) cancer cells were injected to BALB/cJ mice
P2rx7−/− (Generously provided by Niklas Rye Jørgensen, Department of Clinical Biochemistry, Rigshospitalet, Glostrup, Denmark) or to BALB/cJ
P2rx7+/+ litter mate control mice, which were housed in the same environmentally controlled conditions.
P2rx7−/− mice were backcrossed onto the BALB/cJ background as previously described [
51].
In experiments assessing the effect of
P2rx7 gene expression in mammary cancer cells on primary tumour growth and metastatic progression, 1 × 10
4 CTL (
P2rx7+/
+), Crispr#1 or Crispr#2 (
P2rx7−/−) 4T1-derived mammary cancer-cells (see
Section 4.2) in 100 µL of a PBS solution were injected in the mammary fat pad, under isoflurane anaesthesia, of wild-type BALB/cJ mice (Janvier Labs, Saint Berthevin, France).
In experiments assessing the efficacy of P2X7 antagonism, 1 × 104 CTL (P2rx7+/+) cells were injected in the mammary fat pad of wild-type BALB/cJ mice (Janvier Labs, Saint Berthevin, France), and treatments were randomly administrated once tumours reached 80 mm3. Then, 230 µL of 3 mg/mL A437079 or 100 µL of 300 nM AZ10606120 were injected intraperitoneally every two days. Primary tumours were fixed in formalin, included in paraffin, and cut in 5 µm tissue sections. Slides were deparaffinized, rehydrated and heated in citrate buffer pH 6.0 for antigenic retrieval. Immunohistochemistry was performed using a primary antibody anti-P2X7 (APR-008 Alomone Labs Ltd.) and streptavidin-biotin-peroxidase method with diaminobenzidine as the chromogen (Kit LSAB, Dakocytomation, Dako France SAS, Les Ullis, France). Slides were finally counterstained with haematoxylin. Negative controls were obtained by omission of the primary antibody or incubation with an irrelevant antibody.
4.16. Mathematical Model for Assessing Mammary Tumour Growth Depending on Treatments
A Gompertz model was used to describe mammary tumour growth over time. This model included three parameters, i.e., the first-order growth rate constant (kgrowth), the maximum tumour volume (Vmax) and the power coefficient γ. In addition, the effect of treatment (EFF) on tumour volume was estimated as a parameter being 0 without treatment (i.e., before treatment, in both A438079 and AZ10606120 groups, and every time in the vehicle group) and estimated when treatment was administered. Model parameters were estimated using nonlinear mixed-effects modelling. This approach allows description of the inter-subject variability in the population (mean and inter-subject standard deviations) and quantification of the association of factors of variability (referred as covariates) with inter-subject distribution of Gomperz parameters. The influence of treatment condition (A438079 or AZ10606120) was tested as a covariate on both kgrowth and Vmax. This analysis was carried out using MonolixSuite2018 (Lixoft®, Antony, France). The effects of treatment and treatment conditions on each parameter were tested using the likelihood ratio test (LRT).
4.17. Data Presentation and Statistical Analysis
Data are displayed as median (n = number of cells/ independent experiments). Two-way ANOVA followed by a Dunn’s Multiple Comparison Test, Wilcoxon Signed Rank Test and Mann-Whitney rank sum test were used to compare different conditions, as indicated in the figure legends. Statistical significance is indicated as: * p < 0.05; ** p < 0.01 and *** p < 0.001. NS stands for not statistically different.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}