Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer

 ,
,
Abstract
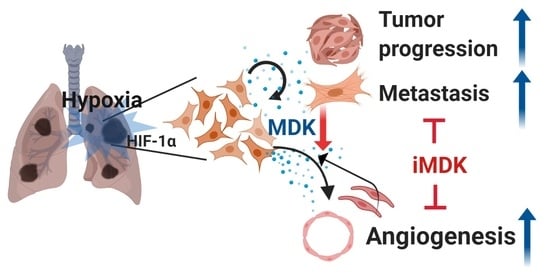
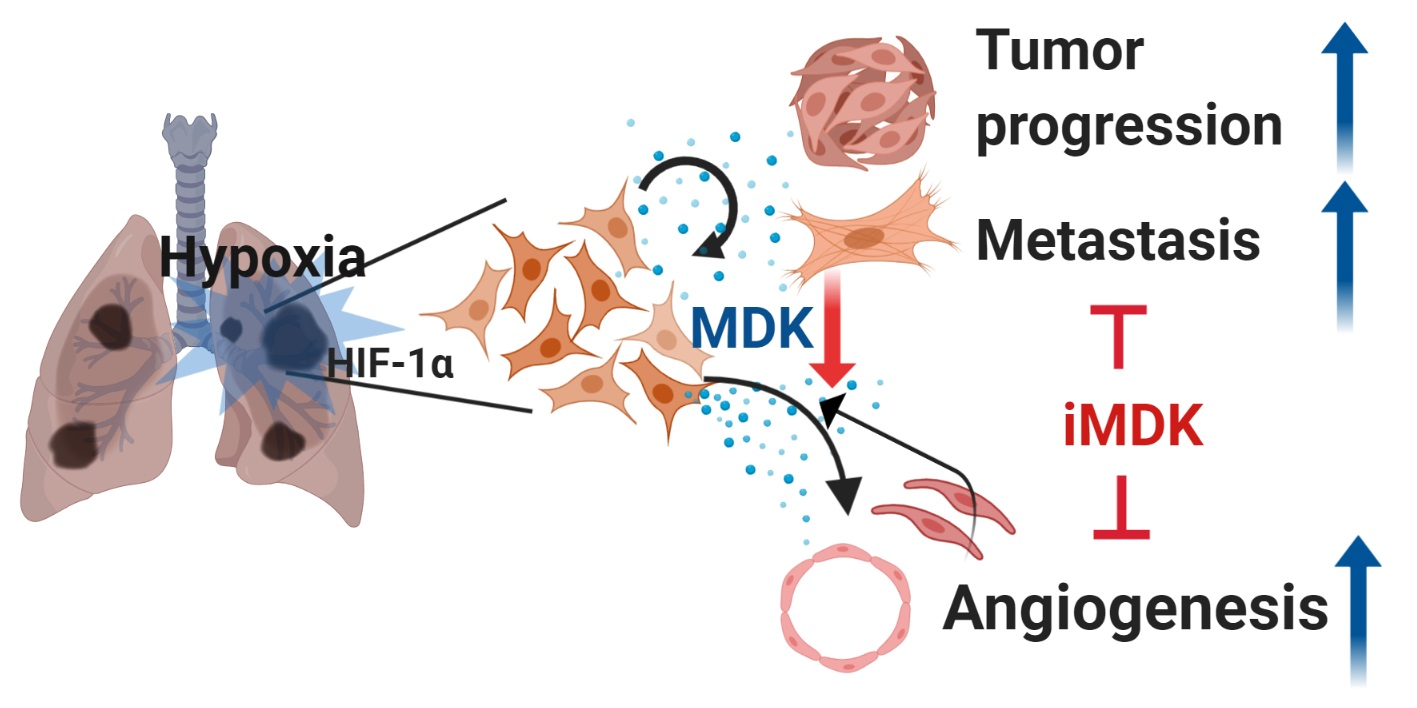
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Lines and Culture Conditions
2.3. Midkine ELISA Assay
2.4. Cell Migration Assay
2.5. Collection of Conditioned Media (CM)
2.6. Cytokine Profiling Antibody Array
2.7. Chick Chorioallantoic Membrane Assay
2.8. Quantitative Real-Time PCR
2.9. Transfection and Establishment of Stable Cell Lines
2.10. Cell Viability and Colony-Forming Assays
2.11. Immunohistochemistry Staining
2.12. Immunoblotting
2.13. Lung Orthotopic Mouse Model
2.14. Tumor Xenografts in Mice and the Spontaneous Lung Metastasis Model
2.15. Statistical Analysis
3. Results
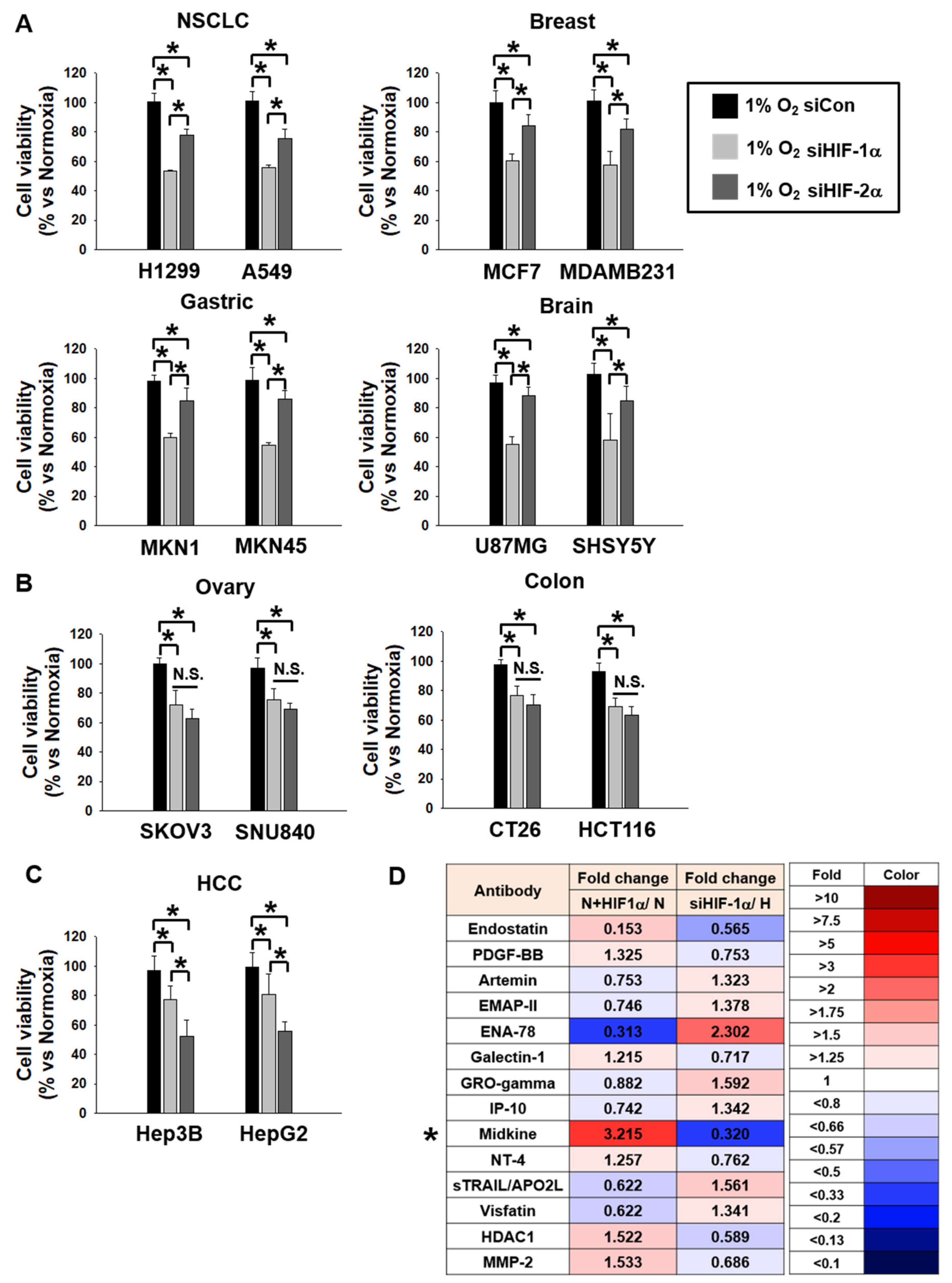
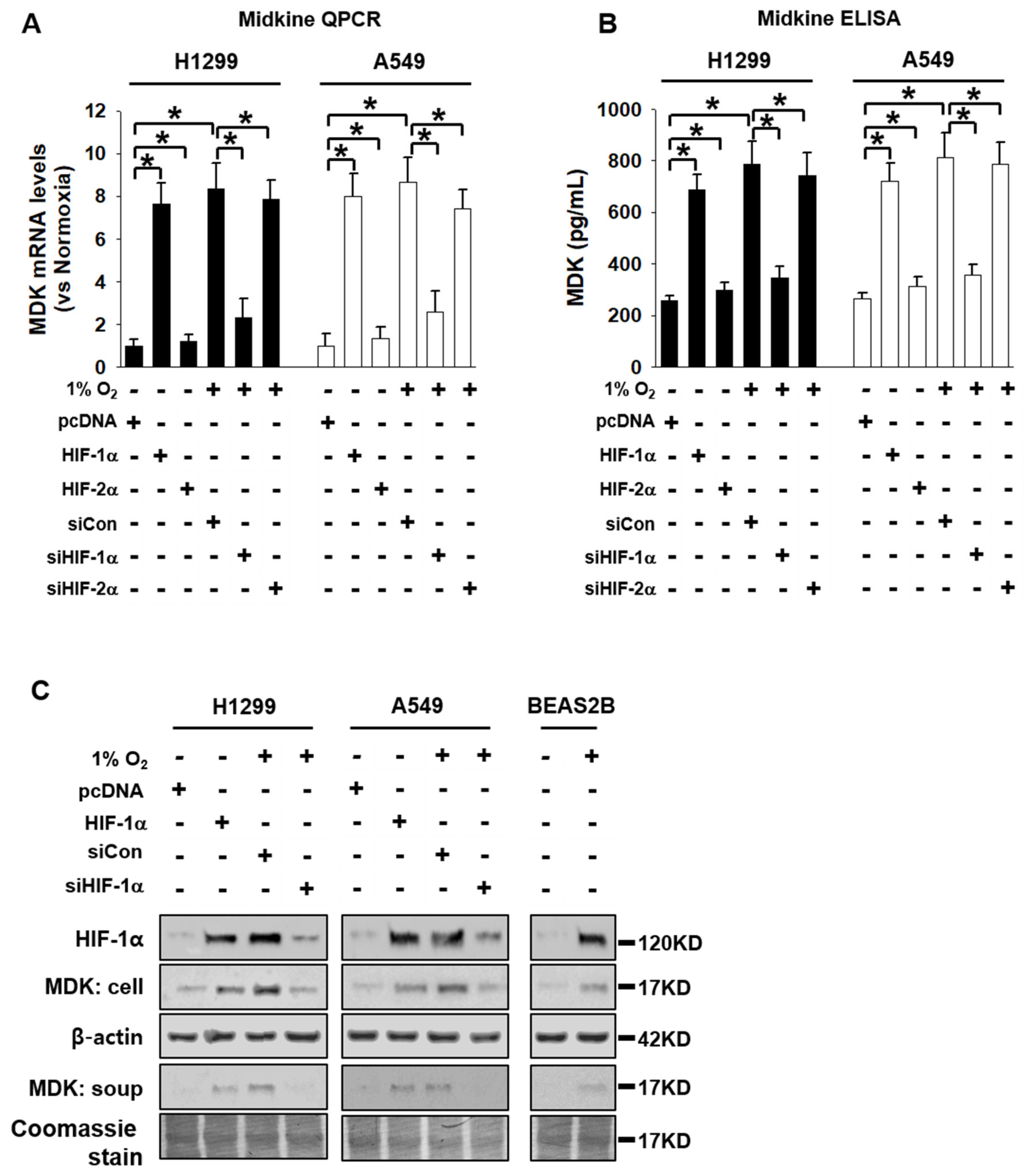
3.1. MDK Expression is Dependent on HIF-1α in NSCLC Cells
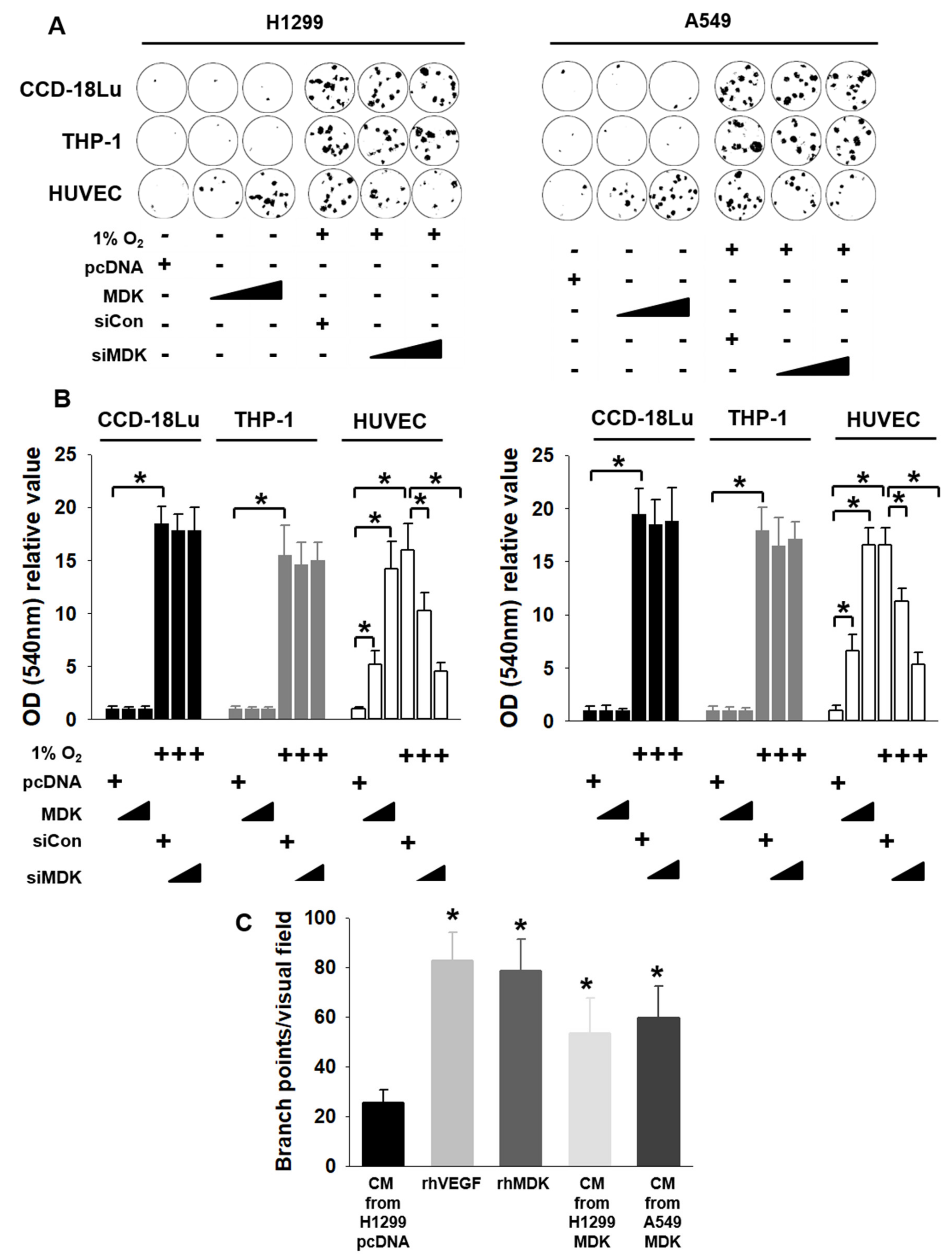
3.2. MDK Promotes the Migration of Endothelial Cells and Angiogenesis
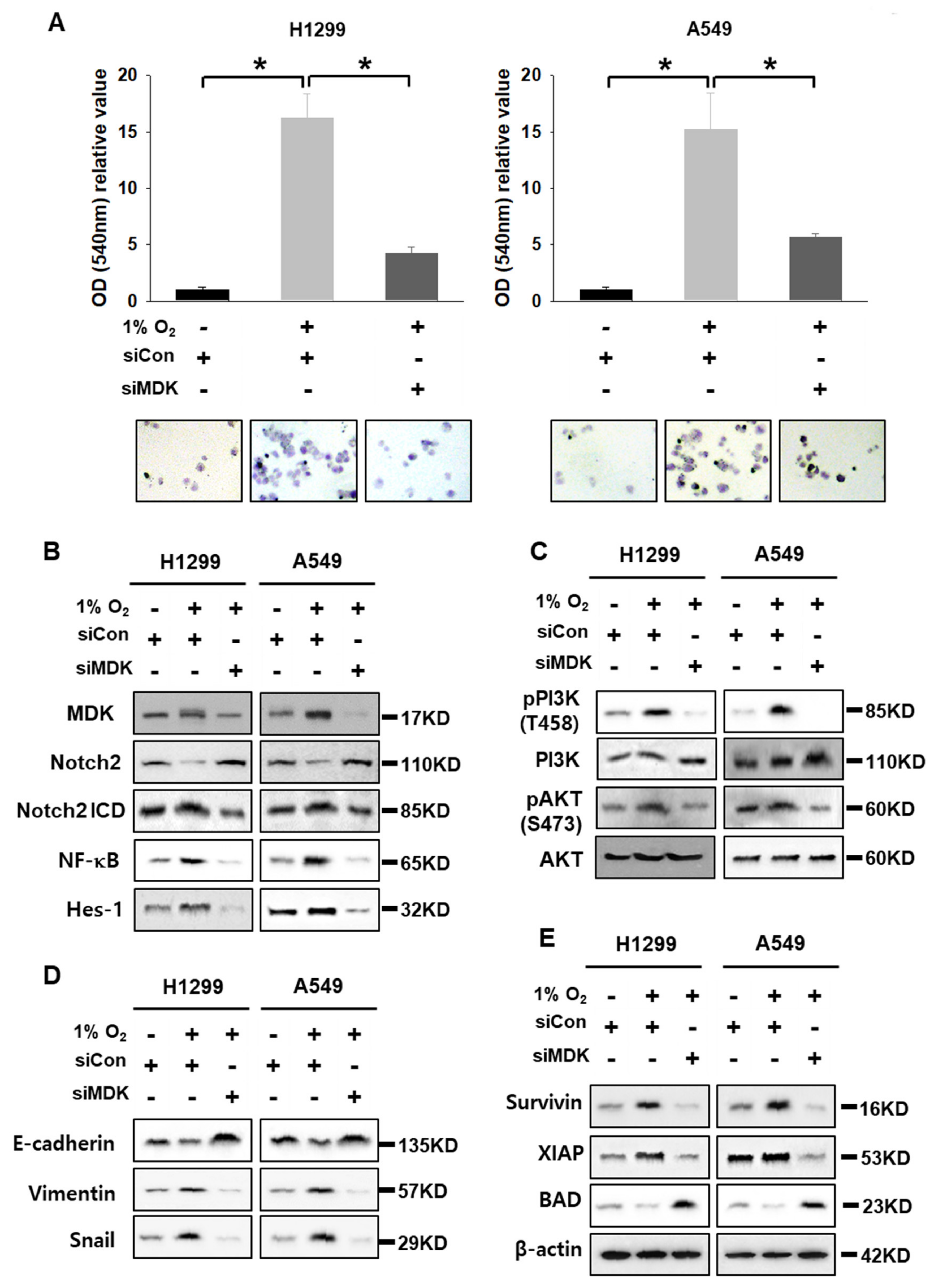
3.3. MDK Overexpression Contributes to EMT in NSCLC Cells
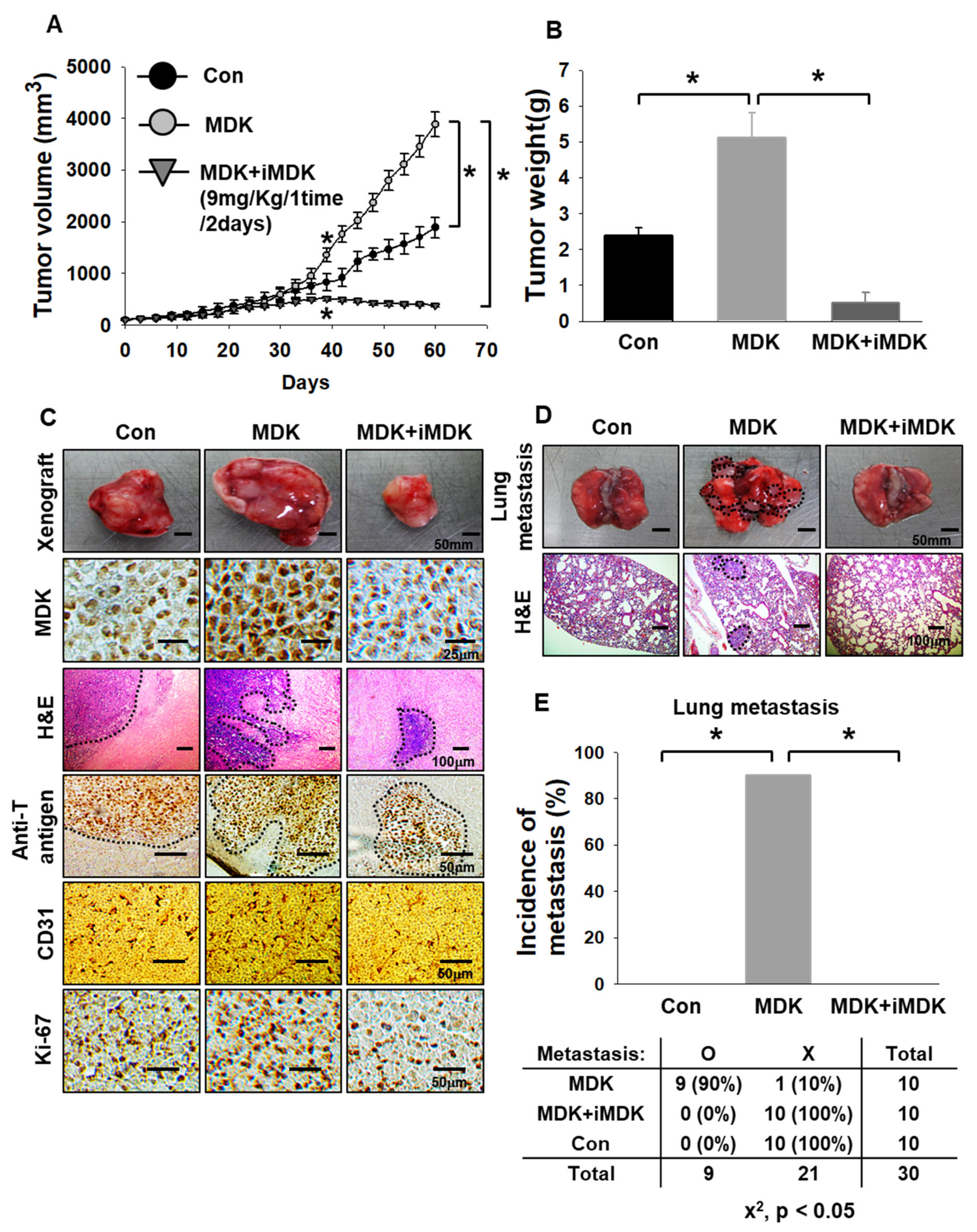
3.4. MDK Inhibitor Exerts Anti-Tumor and Anti-Metastasis Effects in an In Vivo Xenograft Model
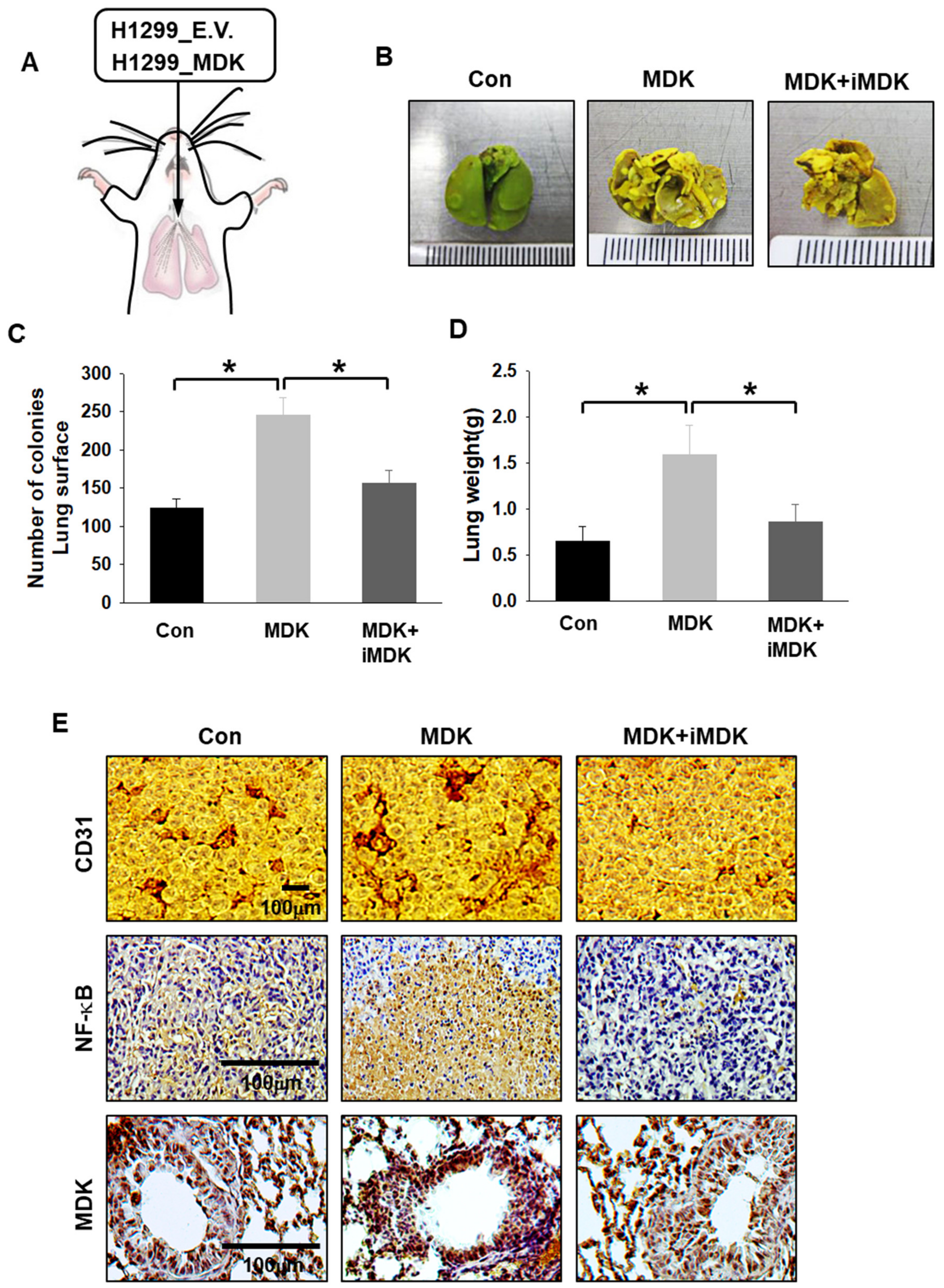
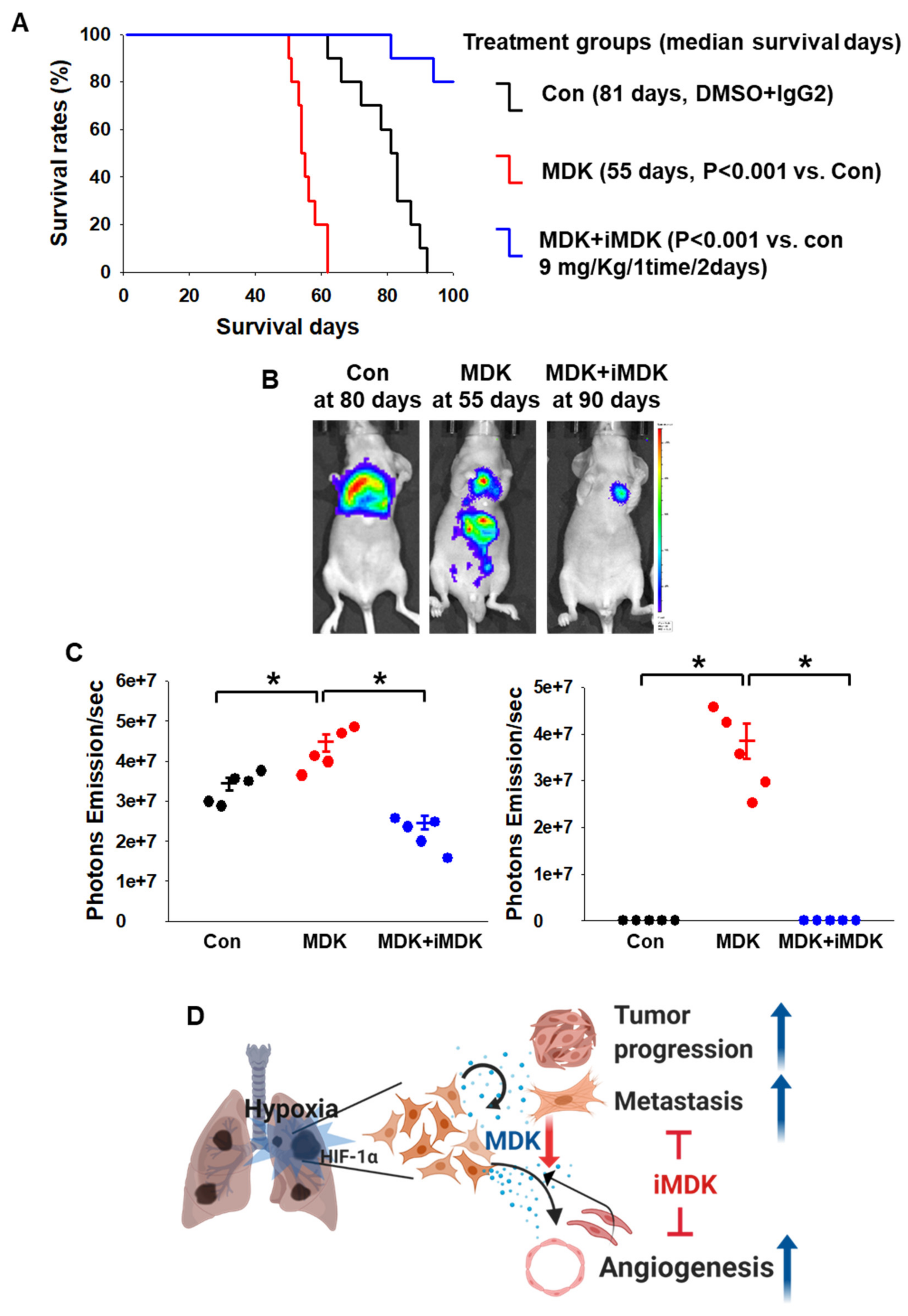
3.5. iMDK Suppresses MDK-Induced Lung Tumor Growth, Angiogenesis, and Metastasis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, J.; Gao, P. Toward normalization of the tumor microenvironment for Cancer Therapy. Integr. Cancer Ther. 2019, 18, 1534735419862352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene 2013, 32, 4057–4063. [Google Scholar] [CrossRef] [Green Version]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar] [PubMed]
- Brizel, D.M.; Scully, S.P.; Harrelson, J.M.; Layfield, L.J.; Bean, J.M.; Prosnitz, L.R.; Dewhirst, M.W. Tumor oxygenation predicts the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996, 56, 941–943. [Google Scholar]
- Osinsky, S.; Zavelevich, M.; Vaupel, P. Tumor hypoxia and malignant progression. Exp. Oncol. 2009, 31, 80–86. [Google Scholar]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basichelix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1, O(2) and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadomatsu, K.; Tomomura, M.; Muramatsu, T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem. Biophys. Res. Commun. 1988, 151, 1312–1318. [Google Scholar] [CrossRef]
- Muramatsu, T. Midkine and pleiotrophin: Two related proteins involved in development, survival, inflammation and tumorigenesis. J. Biochem. 2002, 132, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, H.; Jennbacken, K.; Welen, K.; Damber, J.E. Altered expression of genes regulating angiogenesis in experimental androgen-independent prostate cancer. Prostate 2008, 68, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Muramaki, M.; Miyake, H.; Hara, I.; Kamidono, S. Introduction of midkine gene into human bladder cancer cells enhances their malignant phenotype but increases their sensitivity to antiangiogenic therapy. Clin. Cancer Res. 2003, 9, 5152–5160. [Google Scholar]
- Grupp, K.; Melling, N.; Bogoevska, V.; Reeh, M.; Uzunoglu, F.G.; El Gammal, A.T.; Nentwich, M.F.; Lzbicki, J.R.; Bogoevski, D. Expression of ICAM-1, E-cadherin, periostin and midkine in metastases of pancreatic ductal adenocarcinomas. Exp. Mol. Pathol. 2018, 104, 109–113. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Gungor, C.; Zander, H.; Effenberger, K.E.; Vashist, Y.K.; Kalinina, T.; Izbicki, J.R.; Yekebas, E.; Bockhorm, M. Notch signaling activated by replication stress induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011, 71, 5009–5019. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Hoque, M.O.; Wu, F.; Trink, B.; Sidransky, D.; Ratovitski, E.A. Midkine induces epithelial-mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle 2008, 7, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R. Measuring midkine: The utility of midkine as a biomarker in cancer and other diseases. Br. J. Pharmacol. 2014, 171, 2925–2939. [Google Scholar] [CrossRef] [Green Version]
- Yuan, K.; Chen, Z.; Li, W.; Gao, C.E.; Li, G.; Guo, G.; Yang, Y.; Ai, Y.; Zhang, M. MDK protein overexpression correlates with the malignant status and prognosis of non-small cell lung cancer. Arch. Med. Res. 2015, 46, 635–641. [Google Scholar] [CrossRef]
- Hao, H.; Maeda, Y.; Fukazawa, T.; Takaoka, M.; Bao, X.-H.; Matsuoka, J.; Okui, T.; Shimo, T.; Takigwa, N.; Tomono, Y.; et al. Inhibition of the Growth Factor MDK/midkine by a Novel Small Molecule Compound to Treat Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e71093. [Google Scholar] [CrossRef] [Green Version]
- Masui, M.; Okui, T.; Shimo, T.; Takabatake, K.; Fukazawa, T.; Matsumoto, K.; Kurio, N.; Ibaragi, S.; Namoto, Y.; Nagatsuka, H.; et al. Novel midkine inhibitor iMDK inhibits tumor growth and angiogenesis in oral squamous cell carcinoma. Anticancer Res. 2016, 36, 2775–2781. [Google Scholar] [PubMed]
- Storgard, C.; Mikolon, D.; Stupack, D.G. Angiogenesis assays in the chick CAM. Methods Mol. Biol. 2005, 294, 123–136. [Google Scholar] [PubMed]
- Coliat, P.; Ramolu, L.; Jégu, J.; Gaiddon, C.; Jung, A.C. Pencreach. Constitutive or induced HIF-2 addiction is involved in resistance to anti-EGFR treatment and radiation therapy in HNSCC. Cancers 2019, 11, 1607–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, P.R.; Mucenski, M.L.; Le Cras, T.D.; Nichols, W.C.; Whitsett, J.A. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J. Biol. Chem. 2004, 279, 37124–37132. [Google Scholar] [CrossRef] [Green Version]
- Filippou, P.S.; Karagiannis, G.S.; Constantinidou, A. Midkine (MDK) growth factor: A key player in cancer progression and a promising therapeutic target. Oncogene 2020, 39, 2040–2054. [Google Scholar] [CrossRef]
- Goldfarb, M. The fibroblast growth factor family. Cell Growth Differ. 1990, 1, 439–445. [Google Scholar]
- Ferrara, N.; Houck, K.; Jakeman, L.; Leung, D.W. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr. Rev. 1992, 13, 18–32. [Google Scholar] [CrossRef]
- Ferrara, N. The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res Treat 1995, 36, 127–137. [Google Scholar] [CrossRef]
- Choudhuri, R.; Zhang, H.T.; Donnini, S.; Ziche, M.; Bicknell, R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997, 57, 1814–1819. [Google Scholar]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 versus HIF-2 is one more important than the other? Vascul. Pharmacol. 2012, 56, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Zagorska, A.; Dulak, J. HIF-1: The knowns and unknowns of hypoxia sensing. Acta Biochim. Pol. 2004, 51, 563–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotch, L.E.; Iyer, N.V.; Laughner, E.; Semenza, G.L. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev. Biol. 1999, 209, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, D.-F.; Rui, Y.; Jiang, A.-B.; Liu, Z.-L.; Huang, L.-N. Prognosis value of HIF-1α expression in patients with non-small cell lung cancer. Gene 2014, 541, 69–74. [Google Scholar] [CrossRef]
- Tomomura, M.; Kadomatsu, K.; Matsubara, S.; Muramatsu, T. A retinoic acid-responsive gene, MK, found in the teratocarcinoma system. J. Biol. Chem. 1990, 265, 10765–10770. [Google Scholar]
- Kadomatsu, K.; Huang, R.; Suganuma, T.; Murata, F.; Muramatsu, T. A retinoic acid responsive gene, MK, found in the teratocarcinoma system is expressed in a spatially and temporally controlled manner during mouse embryogenesis. J. Cell Biol. 1990, 110, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Owada, K.; Sanjo, N.; Kobayashi, T.; Mizusawa, H.; Muramatsu, T.; Michikawa, M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J. Neurochem. 1999, 73, 2084–2092. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, D.H.; Jo, J.Y.; Kim, S.H.; Choi, M.; Han, C.; Choi, B.K.; Kim, S.S. Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer. Cancers 2020, 12, 2402. https://doi.org/10.3390/cancers12092402
Shin DH, Jo JY, Kim SH, Choi M, Han C, Choi BK, Kim SS. Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer. Cancers. 2020; 12(9):2402. https://doi.org/10.3390/cancers12092402
Chicago/Turabian StyleShin, Dong Hoon, Jeong Yeon Jo, Sun Ha Kim, Minyoung Choi, Chungyong Han, Beom K. Choi, and Sang Soo Kim. 2020. "Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer" Cancers 12, no. 9: 2402. https://doi.org/10.3390/cancers12092402
APA StyleShin, D. H., Jo, J. Y., Kim, S. H., Choi, M., Han, C., Choi, B. K., & Kim, S. S. (2020). Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer. Cancers, 12(9), 2402. https://doi.org/10.3390/cancers12092402