Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors



 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
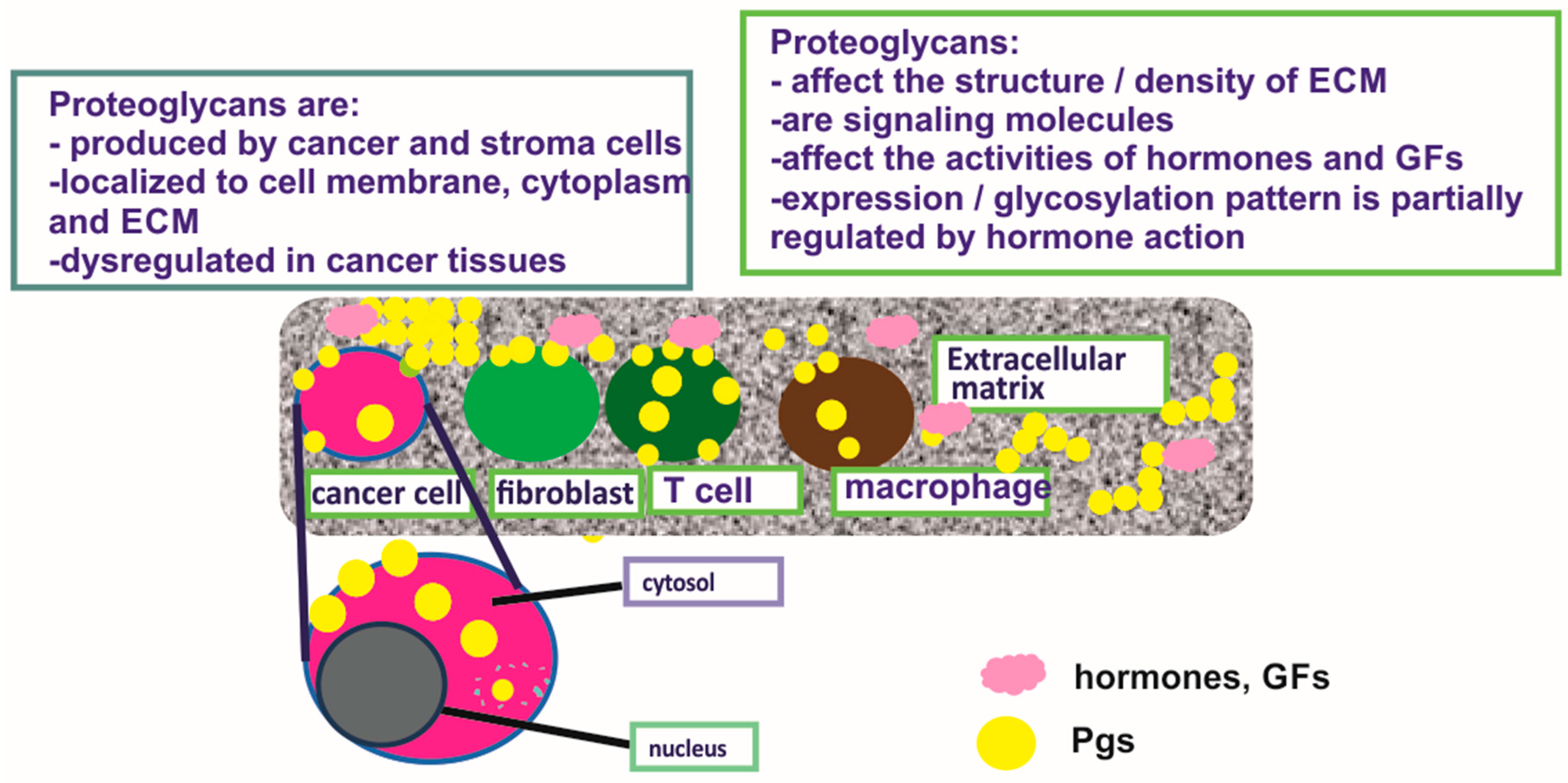
2. Proteoglycans
3. Proteoglycan Expression in Hormone-Dependent Cancer
4. Role of PGs in Hormone-Dependent Cancer Growth, Metastasis, and Angiogenesis
4.1. Effects of PGs on Cancer Cell Growth and Epithelial-to-Mesenchymal Transition (EMT)
4.2. SLRPs—Unique Regulators of Cancer Cell Functions
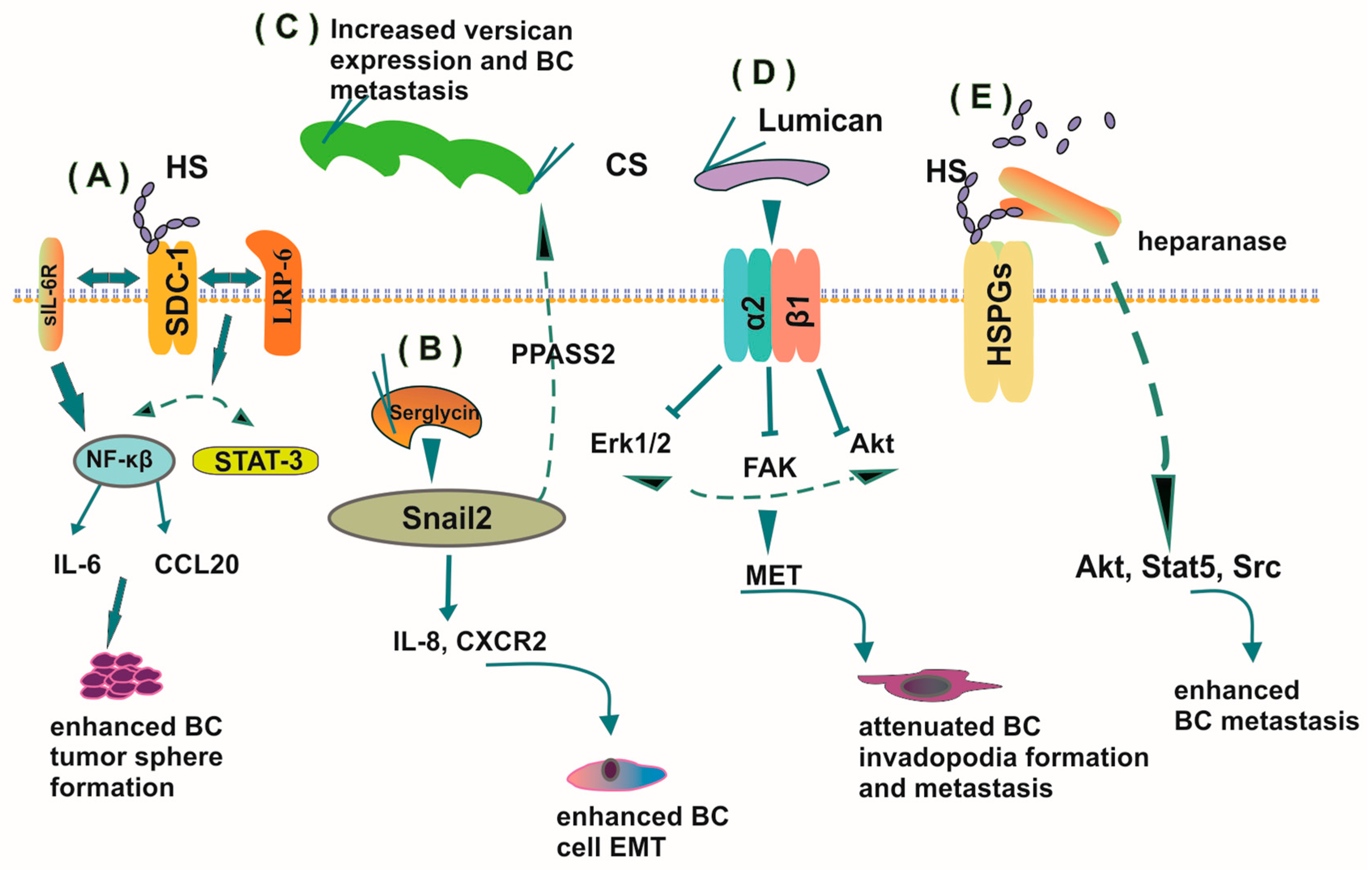
4.3. PGs in the Process of Hormone-Dependent Cancer Metastasis
4.4. The Role of PGs in Tumor-Dependent Angiogenesis
4.5. Xenobiotics and Hormone-Dependent Cancer-Implications of the ECM Niche
5. The Contribution of PGs to the Immunobiology of Hormone-Dependent Cancer
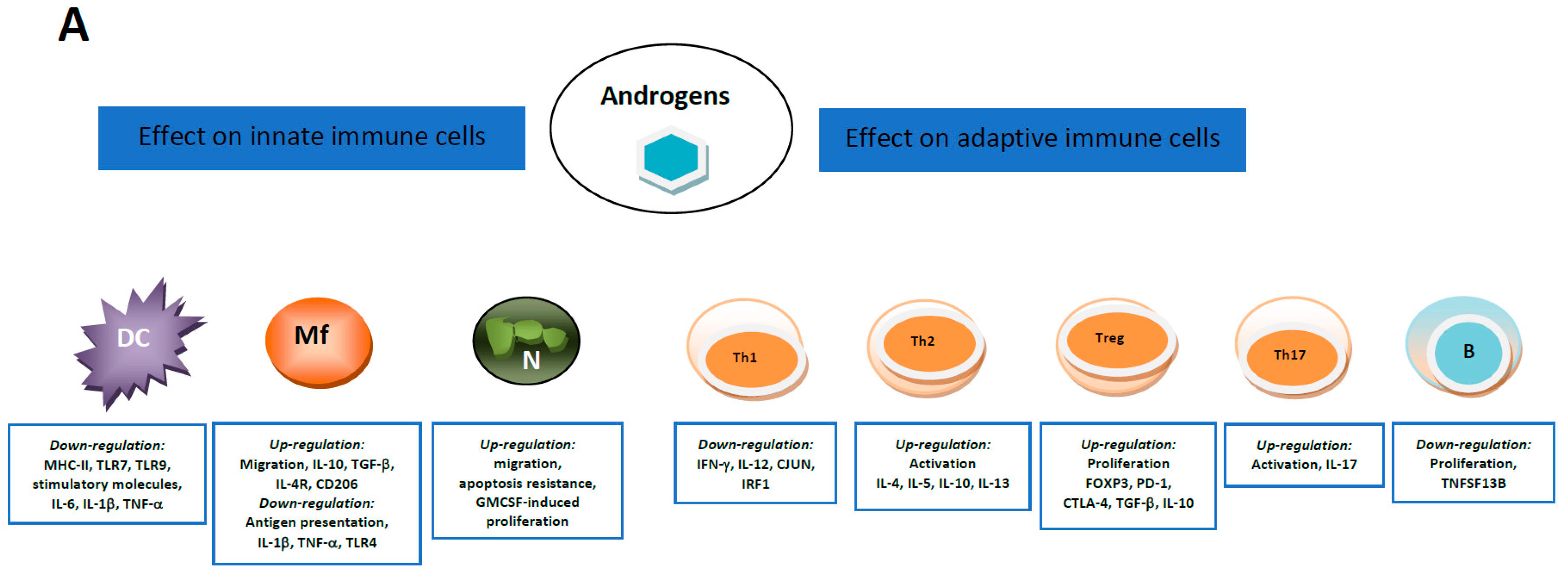
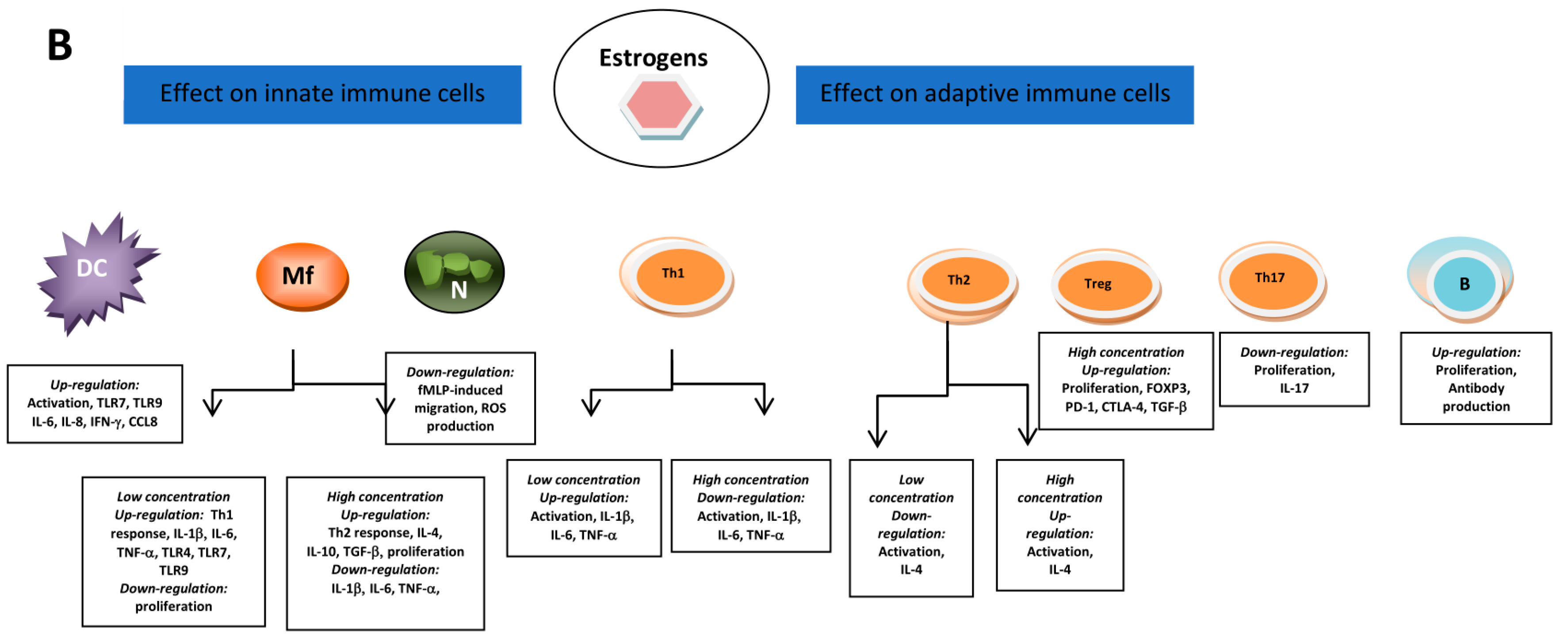
5.1. Androgens and Estrogens Influencers of Immune Cells
5.2. Breast Cancer—Immunological Perspective and Contribution of PGs
5.3. Prostate Cancer-Immunological Perspective
6. Autophagy Affects Chemoresistance in Hormone-Dependent Cancers—Role of PGs
7. Conclusions
Funding
Conflicts of Interest
References
- Yu, Y.-R.; Ho, P.-C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzanakakis, G.; Neagu, M.; Tsatsakis, A.; Nikitovic, D. Proteoglycans and Immunobiology of Cancer—Therapeutic Implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer Microenvironment and Inflammation: Role of Hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastassiades, O.T.; Pryce, D.M. Fibrosis as an Indication of Time in Infiltrating Breast Cancer and its Importance in Prognosis. Br. J. Cancer 1974, 29, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Essentials of Glycobiology. In Proteoglycans and Sulfated Glycosaminoglycans, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2017. [Google Scholar]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; A Carr, S.; O Hynes, R. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014, 3, e01308. [Google Scholar] [CrossRef] [Green Version]
- Nikitovic, D.; Papoutsidakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican affects tumor cell functions, tumor–ECM interactions, angiogenesis and inflammatory response. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 35, 206–214. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Urbonas, T.; A Silva, M.; Muschel, R.J.; Gordon-Weeks, A.N. A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Heyland, A.; Hodin, J.; Reitzel, A.M. Hormone signaling in evolution and development: A non-model system approachs. BioEssays News Rev. Mol. Cell. Dev. Biol. 2005, 27, 64–75. [Google Scholar] [CrossRef]
- Ruhs, S.; Nolze, A.; Hübschmann, R.; Grossmann, C. 30 Years of the Mineralocorticoid Receptor: Nongenomic effects via the mineralocorticoid receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Michalski, S.; Kommagani, R. Role for Growth Regulation by Estrogen in Breast Cancer 1 (GREB1) in Hormone-Dependent Cancers. Int. J. Mol. Sci. 2018, 19, 2543. [Google Scholar] [CrossRef] [Green Version]
- Subramani, R.; Nandy, S.B.; Pedroza, D.A.; Lakshmanaswamy, R. Role of Growth Hormone in Breast Cancer. Endocrinology 2017, 158, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bupp, M.R.G.; Potluri, T.; Fink, A.L.; Klein, S.L. The Confluence of Sex Hormones and Aging on Immunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural Insights into Biological Roles of Protein-Glycosaminoglycan Interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Kolset, S.O.; Tveit, H. Serglycin—Structure and biology. Cell Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Nikitovic, D.; Mytilinaiou, M.; Berdiaki, A.; Karamanos, N.K.; Tzanakakis, G.N. Heparan sulfate proteoglycans and heparin regulate melanoma cell functions. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2471–2481. [Google Scholar] [CrossRef]
- Listik, E.; Gaschler, J.A.M.; Matias, M.; Feres, M.F.N.; Toma, L.; Nahás-Scocate, A.C.R.; Gascheler, J.A.M. Proteoglycans and dental biology: The first review. Carbohydr. Polym. 2019, 225, 115199. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Assouti, M.; Sifaki, M.; Katonis, P.; Krasagakis, K.; Karamanos, N.K.; Tzanakakis, G.N. Chondroitin sulfate and heparan sulfate-containing proteoglycans are both partners and targets of basic fibroblast growth factor-mediated proliferation in human metastatic melanoma cell lines. Int. J. Biochem. Cell Biol. 2008, 40, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Akintayo, A.; Stanley, P. Roles for Golgi Glycans in Oogenesis and Spermatogenesis. Front. Cell Dev. Biol. 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Douaiher, J.; Succar, J.; Lancerotto, L.; Gurish, M.F.; Orgill, D.P.; Hamilton, M.J.; Krilis, S.A.; Stevens, R.L. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv. Immunol. 2014, 122, 211–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpetinou, A.; Skandalis, S.S.; Labropoulou, V.T.; Smirlaki, G.; Noulas, A.; Karamanos, N.K.; Theocharis, A.D. Serglycin: At the Crossroad of Inflammation and Malignancy. Front. Oncol. 2014, 3, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V. MATRIX PROTEOGLYCANS: From Molecular Design to Cellular Function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Toole, B.P.; Hascall, V.C. Hyaluronan and the Aggregating Proteoglycans. In The Extracellular Matrix: An Overview; Mecham, R.P., Ed.; Springer: Berlin, Germany, 2011; pp. 147–195. [Google Scholar]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y. Brevican: A major proteoglycan in adult brain. Perspect. Dev. Neurobiol. 1996, 3, 307–317. [Google Scholar]
- Liu, B.P.; Cafferty, W.B.; O Budel, S.; Strittmatter, S.M. Extracellular regulators of axonal growth in the adult central nervous system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1593–1610. [Google Scholar] [CrossRef]
- Theocharidis, U.; Long, K.; Ffrench-Constant, C.; Faissner, A. Regulation of the neural stem cell compartment by extracellular matrix constituents. Prog. Brain Res. 2014, 214, 3–28. [Google Scholar] [CrossRef]
- Henrich-Noack, P.; Nikitovic, D.; Neagu, M.; Docea, A.O.; Engin, A.B.; Gelperina, S.; Shtilman, M.; Mitsias, P.; Tzanakakis, G.; Gozes, I.; et al. The blood–brain barrier and beyond: Nano-based neuropharmacology and the role of extracellular matrix. Nanomedicine 2019, 17, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R. Transmembrane Signaling Proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114. [Google Scholar] [CrossRef] [PubMed]
- Mytilinaiou, M.; Nikitovic, D.; Berdiaki, A.; Kostouras, A.; Papoutsidakis, A.; Tsatsakis, A.; Tzanakakis, G.N. Emerging roles of syndecan 2 in epithelial and mesenchymal cancer progression. IUBMB Life 2017, 69, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Reiland, J.; Sanderson, R.D.; Waguespack, M.; Barker, S.A.; Long, R.; Carson, D.D.; Marchetti, D. Heparanase Degrades Syndecan-1 and Perlecan Heparan Sulfate: Functional implications for tumor cell invasion. J. Biol. Chem. 2003, 279, 8047–8055. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V.; Murdoch, A.D. Proteoglycans of the extracellular environment: Clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 598–614. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V. The Family of the Small Leucine-Rich Proteoglycans: Key Regulators of Matrix Assembly and Cellular Growth. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 141–174. [Google Scholar] [CrossRef]
- Danielson, K.G.; Fazzio, A.; Cohen, I.; Cannizzaro, L.A.; Eichstetter, I.; Iozzo, R.V. The human decorin gene: Intron-exon organization, discovery of two alternatively spliced exons in the 5′ untranslated region, and mapping of the gene to chromosome 12q23. Genomics 1993, 15, 146–160. [Google Scholar] [CrossRef]
- Nikitovic, D.; Berdiaki, K.; Chalkiadaki, G.; Karamanos, N.K.; Tzanakakis, G. The Role of SLRP-Proteoglycans in Osteosarcoma Pathogenesis. Connect. Tissue Res. 2008, 49, 235–238. [Google Scholar] [CrossRef]
- Schaefer, L.; Iozzo, R.V. Biological Functions of the Small Leucine-rich Proteoglycans: From Genetics to Signal Transduction. J. Biol. Chem. 2008, 283, 21305–21309. [Google Scholar] [CrossRef] [Green Version]
- Nikitovic, D.; Aggelidakis, J.; Young, M.F.; Iozzo, R.V.; Karamanos, N.K.; Tzanakakis, G.N. The Biology of Small Leucine-rich Proteoglycans in Bone Pathophysiology. J. Biol. Chem. 2012, 287, 33926–33933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteoglycan neofunctions: Regulation of inflammation and autophagy in cancer biology. FEBS J. 2016, 284, 10–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafiropoulos, A.; Nikitovic, D.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Decorin-Induced Growth Inhibition Is Overcome through Protracted Expression and Activation of Epidermal Growth Factor Receptors in Osteosarcoma Cells. Mol. Cancer Res. 2008, 6, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voudouri, K.; Nikitovic, D.; Berdiaki, A.; Kletsas, D.; Karamanos, N.K.; Tzanakakis, G.N. IGF-I/EGF and E2 signaling crosstalk through IGF-IR conduit point affects breast cancer cell adhesion. Matrix Biol. J. Int. Soc. Matrix Biol. 2016, 56, 95–113. [Google Scholar] [CrossRef]
- Aggelidakis, J.; Berdiaki, A.; Nikitovic, D.; Papoutsidakis, A.; Papachristou, D.J.; Tsatsakis, A.M.; Tzanakakis, G.N. Biglycan Regulates MG63 Osteosarcoma Cell Growth Through a LPR6/beta-Catenin/IGFR-IR Signaling Axis. Front. Oncol. 2018, 8, 470. [Google Scholar] [CrossRef] [Green Version]
- Papoutsidakis, A.; Giatagana, E.M.; Berdiaki, A.; Spyridaki, I.; Spandidos, D.A.; Tsatsakis, A.; Tzanakakis, G.N.; Nikitovic, D. Lumican mediates HTB94 chondrosarcoma cell growth via an IGF-IR/Erk1/2 axis. Int. J. Oncol. 2020, in press. [Google Scholar]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Grozescu, T.; Popa, F. Prostate cancer between prognosis and adequate/proper therapy. J. Med. Life 2017, 10, 5–12. [Google Scholar]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef]
- Suhovskih, A.V.; Mostovich, L.A.; Kunin, I.S.; Boboev, M.M.; Nepomnyashchikh, G.I.; Aidagulova, S.V.; Grigorieva, E.V. Proteoglycan Expression in Normal Human Prostate Tissue and Prostate Cancer. ISRN Oncol. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Farfán, N.; Ocarez, N.; Castellón, E.A.; Mejía, N.; De Herreros, A.G.; Contreras, H.R. The transcriptional factor ZEB1 represses Syndecan 1 expression in prostate cancer. Sci. Rep. 2018, 8, 11467. [Google Scholar] [CrossRef] [Green Version]
- Kiviniemi, J.; Kallajoki, M.; Kujala, I.; Matikainen, M.-T.; Alanen, K.; Jalkanen, M.; Salmivirta, M. Altered expression of syndecan-1 in prostate cancer. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2004, 112, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Popovic, A.; Demirović, A.; Spajić, B.; Štimac, G.; Krušlin, B.; Tomas, D. Expression and prognostic role of syndecan-2 in prostate cancer. Prostate Cancer Prostatic Dis. 2009, 13, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledezma, R.; Cifuentes, F.; Gallegos, I.; Fullá, J.; Ossandón, E.; A Castellón, E.; Contreras, H.R. Altered expression patterns of syndecan-1 and -2 predict biochemical recurrence in prostate cancer. Asian J. Androl. 2011, 13, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; De Paula, C.A.A.; Carneiro, C.R.; Ortiz, V.; Toma, L.; Kao, W.W.-Y.; Nader, H.B. Lumican expression, localization and antitumor activity in prostate cancer. Exp. Cell Res. 2013, 319, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettin, A.; Reyes, I.; Reyes, N. Gene Expression Profiling of Prostate Cancer–Associated Genes Identifies Fibromodulin as Potential Novel Biomarker for Prostate Cancer. Int. J. Biol. Markers 2016, 31, 153–162. [Google Scholar] [CrossRef]
- Reyes, N.; Benedetti, I.; Bettin, A.; Rebollo, J.; Geliebter, J. The small leucine rich proteoglycan fibromodulin is overexpressed in human prostate epithelial cancer cell lines in culture and human prostate cancer tissue. Cancer Biomark. Sect. A Dis. Markers 2016, 16, 191–202. [Google Scholar] [CrossRef]
- Campbell, D.H.; Lund, M.E.; Nocon, A.L.; Cozzi, P.J.; Frydenberg, M.; De Souza, P.; Schiller, B.; Beebe-Dimmer, J.L.; Ruterbusch, J.J.; Walsh, B. Detection of glypican-1 (GPC-1) expression in urine cell sediments in prostate cancer. PLoS ONE 2018, 13, e0196017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, Z.; Wang, L.; Qiao, B.; Du, E.; Li, L.; Xu, Y.; Zhang, Z. Prognostic significance of GPC5 expression in patients with prostate cancer. Tumor Biol. 2015, 37, 6413–6418. [Google Scholar] [CrossRef]
- Terry, D.E.; Clark, A.F. Influence of testosterone on chondroitin sulphate proteoglycan in the rat prostate. Biochem. Cell Biol. 1996, 74, 645–651. [Google Scholar] [CrossRef]
- Kofoed, J.A.; Tumilasci, O.R.; Curbelo, H.M.; Lemos, S.M.F.; Arias, N.H.; Houssay, A.B. Effects of castration and androgens upon prostatic proteoglycans in rats. Prostate 1990, 16, 93–102. [Google Scholar] [CrossRef]
- Scott, E.; Munkley, J. Glycans as Biomarkers in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, I.J. Proteoglycans in prostate cancer. Nat. Rev. Urol. 2012, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Gesteira, T.F.; Coulson-Thomas, Y.M.; Vicente, C.M.; Tersariol, I.L.S.; Nader, H.B.; Toma, L. Fibroblast and prostate tumor cell cross-talk: Fibroblast differentiation, TGF-β, and extracellular matrix down-regulation. Exp. Cell Res. 2010, 316, 3207–3226. [Google Scholar] [CrossRef] [PubMed]
- Suhovskih, A.V.; Kashuba, V.I.; Klein, G.; Grigorieva, E.V. Prostate cancer cells specifically reorganize epithelial cell-fibroblast communication through proteoglycan and junction pathways. Cell Adhes. Migr. 2016, 11, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Neill, T.; Multhaupt, H.A.B.; Hubo, M.; Frey, H.; Gopal, S.; Gómes, A.; Afratis, N.; Lim, H.C.; et al. Insights into the key roles of proteoglycans in breast cancer biology and translational medicine. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1855, 276–300. [Google Scholar] [CrossRef] [Green Version]
- Hanna, M.; Diorio, C. Does mammographic density reflect the expression of breast cancer markers? Climacteric J. Int. Menopause Soc. 2013, 16, 407–416. [Google Scholar] [CrossRef]
- Britt, K.; Ingman, W.; Huo, C.; Chew, G.; Thompson, E. The pathobiology of mammographic density. J. Cancer Biol Res. 2014, 2, 1021–1031. [Google Scholar]
- Shawky, M.S.; Ricciardelli, C.; Lord, M.S.; Whitelock, J.; Ferro, V.; Britt, K.L.; Thompson, E.W. Proteoglycans: Potential Agents in Mammographic Density and the Associated Breast Cancer Risk. J. Mammary Gland. Biol. Neoplasia 2015, 20, 121–131. [Google Scholar] [CrossRef]
- Jansson, M.; Billing, O.; Herdenberg, C.; Lundin, C.; Tolockiene, E.; Nazemroaya, A.; Sund, M. Expression and Circulating Levels of Perlecan in Breast Cancer—Implications for Oestrogen Dependent Stromal Remodeling. J. Mammary Gland. Biol. Neoplasia 2020, 25, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Stoeckelhuber, M.; Stumpf, P.; Hoefter, E.A.; Welsch, U. Proteoglycan–collagen associations in the non-lactating human breast connective tissue during the menstrual cycle. Histochem. Cell Biol. 2002, 118, 221–230. [Google Scholar] [CrossRef]
- Lteif, A.; Javed, A. Development of the Human Breast. Semin. Plast. Surg. 2013, 27, 005–012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löfgren, L.; Sahlin, L.; Jiang, S.; Von Schoultz, B.; Fernstad, R.; Skoog, L.; Von Schoultz, E. Expression of syndecan-1 in paired samples of normal and malignant breast tissue from postmenopausal women. Anticancer Res. 2007, 27, 3045–3050. [Google Scholar] [PubMed]
- Maeda, T.; Alexander, C.M.; Friedl, A. Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Res. 2004, 64, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundström, E.; Sahlin, L.; Skoog, L.; Hagerstrom, T.; Svane, G.; Azavedo, E.; Sandelin, K.; Von Schoultz, B. Expression of syndecan-1 in histologically normal breast tissue from postmenopausal women with breast cancer according to mammographic density. Climacteric J. Int. Menopause Soc. 2006, 9, 277–282. [Google Scholar] [CrossRef]
- Hallberg, G.; Andersson, E.; Naessén, T.; Ekman-Ordeberg, G. The expression of syndecan-1, syndecan-4 and decorin in healthy human breast tissue during the menstrual cycle. Reprod. Biol. Endocrinol. 2010, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.C.; Multhaupt, H.A.B.; Couchman, J.R. Cell surface heparan sulfate proteoglycans control adhesion and invasion of breast carcinoma cells. Mol. Cancer 2015, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.C.; Couchman, J.R. Syndecan-2 regulation of morphology in breast carcinoma cells is dependent on RhoGTPases. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2482–2490. [Google Scholar] [CrossRef]
- Kousidou, O.C.; Berdiaki, A.; Kletsas, D.; Zafiropoulos, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N. Estradiol–estrogen receptor: A key interplay of the expression of syndecan-2 and metalloproteinase-9 in breast cancer cells. Mol. Oncol. 2008, 2, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Maruyama, H.; Guo, F.; Kleeff, J.; Itakura, J.; Matsumoto, Y.; Lander, A.D.; Korc, M. Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res. 2001, 61, 5562–5569. [Google Scholar]
- Xiang, Y.-Y.; Ladeda, V.; Filmus, J. Glypican-3 expression is silenced in human breast cancer. Oncogene 2001, 20, 7408–7412. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.G.; Farías, E.; Colombo, L.; Filmus, J.; Puricelli, L.; Joffé, E.B.D.K. Inhibition of Invasion and Metastasis by Glypican-3 in a Syngeneic Breast Cancer Model. Breast Cancer Res. Treat. 2003, 80, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, K.; Seliger, B. Tumor-dependent Effects of Proteoglycans and Various Glycosaminoglycan Synthesizing Enzymes and Sulfotransferases on Patients’ Outcome. Curr. Cancer Drug Targets 2019, 19, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, S.; Embree, M.C.; Bi, Y.; Young, M.F. Regulation, Regulatory Activities, and Function of Biglycan. Crit. Rev. Eukaryot. Gene Expr. 2004, 14, 16. [Google Scholar] [CrossRef]
- Van Bockstal, M.; Lambein, K.; Van Gele, M.; De Vlieghere, E.; Limame, R.; Braems, G.; Van den Broecke, R.; Cocquyt, V.; Denys, H.; Bracke, M.; et al. Differential regulation of extracellular matrix protein expression in carcinoma-associated fibroblasts by TGF-beta1 regulates cancer cell spreading but not adhesion. Oncoscience 2014, 1, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leygue, E.; Snell, L.; Dotzlaw, H.; Troup, S.; Hiller-Hitchcock, T.; Murphy, L.C.; Roughley, P.J.; Watson, P.H. Lumican and decorin are differentially expressed in human breast carcinoma. J. Pathol. 2000, 192, 313–320. [Google Scholar] [CrossRef]
- Leygue, E.; Snell, L.; Dotzlaw, H.; Hole, K.; Hiller-Hitchcock, T.; Roughley, P.J.; Watson, P.H.; Murphy, L.C. Expression of lumican in human breast carcinoma. Cancer Res. 1998, 58, 1348–1352. [Google Scholar]
- Alowami, S.; Troup, S.; Al-Haddad, S.; Kirkpatrick, I.; Watson, P.H. Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res. 2003, 5, R129–R135. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Couch, F.J.; Ahmed, S.; Dunning, A.M.; Pharoah, P.D.; Easton, D.; Fredericksen, Z.; Vierkant, R.A.; Pankratz, V.S.; Goode, E.L.; et al. Genetic variation in stromal proteins decorin and lumican with breast cancer: Investigations in two case-control studies. Breast Cancer Res. 2008, 10, R98. [Google Scholar] [CrossRef] [Green Version]
- Ricciardelli, C.; Sakko, A.J.; Ween, M.P.; Russell, D.L.; Horsfall, D.J. The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev. 2009, 28, 233–245. [Google Scholar] [CrossRef]
- Kischel, P.; Waltregny, D.; Dumont, B.; Turtoi, A.; Greffe, Y.; Kirsch, S.; De Pauw, E.; Castronovo, V. Versican overexpression in human breast cancer lesions: Known and new isoforms for stromal tumor targeting. Int. J. Cancer 2010, 126, 640–650. [Google Scholar] [CrossRef]
- Canavese, G.; Candelaresi, G.; Castellano, I.; Mano, M. Expression of proteoglycan versican in in situ breast lesions: Relations between stromal changes, histotype, and invasion. Pathol. Res. Pract. 2011, 207, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Eshchenko, T.Y.; Rykova, V.I.; Chernakov, A.E.; Sidorov, S.V.; Grigorieva, E.V. Expression of different proteoglycans in human breast tumors. Biochem. Mosc. 2007, 72, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, D.L.; Katakam, S.K.; Greve, B.; Jang, B.; Oh, E.-S.; Alaniz, L.; Götte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afratis, N.; Karamanou, K.; Piperigkou, Z.; Vynios, D.H.; Theocharis, A.D. The role of heparins and nano-heparins as therapeutic tool in breast cancer. Glycoconj. J. 2016, 34, 299–307. [Google Scholar] [CrossRef]
- Johnson, C.E.; Crawford, B.E.; Stavridis, M.; Ten Dam, G.; Wat, A.L.; Rushton, G.; Ward, C.M.; Wilson, V.; van Kuppevelt, T.H.; Esko, J.D.; et al. Essential alterations of heparan sulfate during the differentiation of embryonic stem cells to Sox1-enhanced green fluorescent protein-expressing neural progenitor cells. Stem Cells 2007, 25, 1913–1923. [Google Scholar] [CrossRef]
- Sasaki, N.; Okishio, K.; Ui-Tei, K.; Saigo, K.; Kinoshita-Toyoda, A.; Toyoda, H.; Nishimura, T.; Suda, Y.; Hayasaka, M.; Hanaoka, K.; et al. Heparan Sulfate Regulates Self-renewal and Pluripotency of Embryonic Stem Cells. J. Biol. Chem. 2007, 283, 3594–3606. [Google Scholar] [CrossRef] [Green Version]
- Kraushaar, D.C.; Dalton, S.; Wang, L. Heparan sulfate: A key regulator of embryonic stem cell fate. Biol. Chem. 2013, 394, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Qiang, W.; Liu, Z.; Serna, V.A.; Druschitz, S.A.; Liu, Y.; Espona, M.; Wei, J.-J.; Kurita, T. Down-regulation of miR-29b is essential for pathogenesis of uterine leiomyoma. Endocrinology 2014, 155, 663–669. [Google Scholar] [CrossRef]
- Uchiumi, K.; Tsuboi, K.; Sato, N.; Ito, T.; Hirakawa, H.; Niwa, T.; Yamaguchi, Y.; Hayashi, S.-I. Cancer stem-like properties of hormonal therapy-resistant breast cancer cells. Breast Cancer 2019, 26, 459–470. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.A.; Hassan, H.; Vilardo, L.; Kumar, S.K.; Kumar, A.V.; Kelsch, R.; Schneider, C.; Kiesel, L.; Eich, H.T.; Zucchi, I.; et al. Syndecan-1 (CD138) Modulates Triple-Negative Breast Cancer Stem Cell Properties via Regulation of LRP-6 and IL-6-Mediated STAT3 Signaling. PLoS ONE 2013, 8, e85737. [Google Scholar] [CrossRef] [PubMed]
- Bouris, P.; Manou, D.; Sopaki-Valalaki, A.; Kolokotroni, A.; Moustakas, A.; Kapoor, A.; Iozzo, R.V.; Karamanos, N.K.; Theocharis, A.D. Serglycin promotes breast cancer cell aggressiveness: Induction of epithelial to mesenchymal transition, proteolytic activity and IL-8 signaling. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 74, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Contreras, H.R.; Ledezma, R.A.; Vergara, J.; Cifuentes, F.; Barra, C.; Cabello, P.; Gallegos, I.; Morales, B.; Huidobro, C.; Castellón, E.A. The expression of syndecan-1 and -2 is associated with Gleason score and epithelial-mesenchymal transition markers, E-cadherin and β-catenin, in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2010, 28, 534–540. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, K.; He, M.; Fan, G.; Lu, H. Overexpression of Glypican 5 (GPC5) Inhibits Prostate Cancer Cell Proliferation and Invasion via Suppressing Sp1-Mediated EMT and Activation of Wnt/beta-Catenin Signaling. Oncol. Res. 2018, 26, 565–572. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Piperigkou, Z.; Perreau, C.; Maquart, F.-X.; Vynios, D.H.; Brézillon, S. Lumican effectively regulates the estrogen receptors-associated functional properties of breast cancer cells, expression of matrix effectors and epithelial-to-mesenchymal transition. Sci. Rep. 2017, 7, 45138. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brézillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef]
- Kocbek, V.; Hevir-Kene, N.; Bersinger, N.A.; Mueller, M.D.; Rizner, T.L. Increased levels of biglycan in endometriomas and peritoneal fluid samples from ovarian endometriosis patients. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2014, 30, 520–524. [Google Scholar] [CrossRef]
- Jacobsen, F.; Kraft, J.; Schroeder, C.; Hube-Magg, C.; Kluth, M.; Lang, D.S.; Simon, R.; Sauter, G.; Izbicki, J.R.; Clauditz, T.S.; et al. Up-regulation of Biglycan is Associated with Poor Prognosis and PTEN Deletion in Patients with Prostate Cancer. Neoplasia 2017, 19, 707–715. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Hu, L.; Duan, Y.-T.; Li, J.-F.; Su, L.-P.; Yan, M.; Zhu, Z.-G.; Liu, B.; Yang, Q.-M. Biglycan enhances gastric cancer invasion by activating FAK signaling pathway. Oncotarget 2014, 5, 1885–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimura, K.; Sudhir, K.; Nigro, J.; Ling, S.; Williams, M.R.I.; Komesaroff, P.; Little, P.J. Androgens Stimulate Human Vascular Smooth Muscle Cell Proteoglycan Biosynthesis and Increase Lipoprotein Binding. Endocrinology 2005, 146, 2085–2090. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Cao, J.; Tian, L.; Shen, Y.; Yang, X.; Lin, Q.; Zhang, R.; Liu, H.; Du, X.; Shi, J.; et al. Aromatase-induced endogenous estrogen promotes tumour metastasis through estrogen receptor-alpha/matrix metalloproteinase 12 axis activation in castration-resistant prostate cancer. Cancer Lett. 2019, 467, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, J.; Geliebter, J.; Reyes, N. ESM-1 siRNA Knockdown Decreased Migration and Expression of CXCL3 in Prostate Cancer Cells. Int. J. Biomed. Sci. IJBS 2017, 13, 35–42. [Google Scholar] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar]
- Zhang, C.; Soori, M.; Miles, F.L.; Sikes, R.A.; Carson, D.D.; Chung, L.W.; Farach-Carson, M.C. Paracrine factors produced by bone marrow stromal cells induce apoptosis and neuroendocrine differentiation in prostate cancer cells. Prostate 2010, 71, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.R.; Grindel, B.J.; Francis, L.; Carson, D.D.; Farach-Carson, M.C. Transcriptional activation by NFkappaB increases perlecan/HSPG2 expression in the desmoplastic prostate tumor microenvironment. J. Cell. Biochem. 2014, 115, 1322–1333. [Google Scholar] [CrossRef] [Green Version]
- Grindel, B.; Li, Q.; Arnold, R.; Petros, J.; Zayzafoon, M.; Muldoon, M.; Stave, J.; Chung, L.W.K.; Farach-Carson, M.C. Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: Tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget 2016, 7, 10433–10447. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired Angiogenesis, Delayed Wound Healing and Retarded Tumor Growth in Perlecan Heparan Sulfate-Deficient Mice. Cancer Res. 2004, 64, 4699–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, L.A.; Tellman, T.V.; Farach-Carson, M.C. Flipping the Molecular Switch: Influence of Perlecan and Its Modifiers in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 133–146. [Google Scholar] [PubMed]
- Cook, L.M.; Frieling, J.S.; Nerlakanti, N.; McGuire, J.J.; Stewart, P.A.; Burger, K.L.; Cleveland, J.L.; Lynch, C. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene 2019, 38, 6959–6969. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Yang, W.; Yee, A.J.M. Roles of versican in cancer biology—Tumorigenesis, progression and metastasis. Histol. Histopathol. 2013, 28, 701–713. [Google Scholar]
- Du, W.W.; Fang, L.; Yang, W.; Sheng, W.; Zhang, Y.; Seth, A.; Yang, B.B.; Yee, A.J.M. The role of versican G3 domain in regulating breast cancer cell motility including effects on osteoblast cell growth and differentiation in vitro—Evaluation towards understanding breast cancer cell bone metastasis. BMC Cancer 2012, 12, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zou, X.; Qian, W.; Weng, X.; Zhang, L.; Zhang, L.; Wang, S.; Cao, X.; Ma, L.; Wei, G.; et al. Enhanced PAPSS2/VCAN sulfation axis is essential for Snail-mediated breast cancer cell migration and metastasis. Cell Death Differ. 2019, 26, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, S.E.; Perez, D.; Pan, T.-C.; Sarkisian, C.J.; Portocarrero, C.P.; Sterner, C.J.; Notorfrancesco, K.L.; Cardiff, R.D.; Chodosh, L.A. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005, 8, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Dos Reis, D.C.; Damasceno, K.A.; De Campos, C.B.; Veloso, E.S.; Pêgas, G.R.A.; Kraemer, L.R.; Rodrigues, M.A.; Mattos, M.S.; Gomes, D.A.; Campos, P.P.; et al. Versican and Tumor-Associated Macrophages Promotes Tumor Progression and Metastasis in Canine and Murine Models of Breast Carcinoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2016, 284, 42–55. [Google Scholar] [CrossRef]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage Activation by Heparanase Is Mediated by TLR-2 and TLR-4 and Associates With Plaque Progression. Arter. Thromb. Vasc. Biol. 2013, 33. [Google Scholar] [CrossRef] [Green Version]
- Boyango, I.; Barash, U.; Fux, L.; Naroditsky, I.; Ilan, N.; Vlodavsky, I. Targeting heparanase to the mammary epithelium enhances mammary gland development and promotes tumor growth and metastasis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 65, 91–103. [Google Scholar] [CrossRef]
- Kyriakopoulou, K.; Kefali, E.; Piperigkou, Z.; Bassiony, H.; Karamanos, N.K. Advances in targeting epidermal growth factor receptor signaling pathway in mammary cancer. Cell. Signal. 2018, 51, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Voudouri, K.; Berdiaki, A.; Tzardi, M.; Tzanakakis, G.N.; Nikitovic, D. Insulin-Like Growth Factor and Epidermal Growth Factor Signaling in Breast Cancer Cell Growth: Focus on Endocrine Resistant Disease. Anal. Cell. Pathol. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrione, A.; Neill, T.; Iozzo, R.V. Dichotomy of decorin activity on the insulin-like growth factor-I system. FEBS J. 2013, 280, 2138–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayyad, M.R.; Puchalapalli, M.; Vergara, N.G.; Wangensteen, S.M.; Moore, M.; Mu, L.; Edwards, C.; Anderson, A.; Kall, S.; Sullivan, M.; et al. Syndecan-1 facilitates breast cancer metastasis to the brain. Breast Cancer Res. Treat. 2019, 178, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Fleurot, E.; Goudin, C.; Hanoux, V.; Bonnamy, P.-J.; Levallet, J. Estrogen receptor-α regulates the expression of syndecan-1 in human breast carcinoma cells. Endocr.-Relat. Cancer 2019, 26, 615–628. [Google Scholar] [CrossRef]
- Mennerich, D.; Vogel, A.; Klaman, I.; Dahl, E.; Lichtner, R.; Rosenthal, A.; Pohlenz, H.-D.; Thierauch, K.-H.; Sommer, A. Shift of syndecan-1 expression from epithelial to stromal cells during progression of solid tumours. Eur. J. Cancer 2004, 40, 1373–1382. [Google Scholar] [CrossRef]
- Hassan, H.; Greve, B.; Pavao, M.S.; Kiesel, L.; Ibrahim, S.A.; Gotte, M. Syndecan-1 modulates beta-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. FEBS J. 2013, 280, 2216–2227. [Google Scholar] [CrossRef]
- Nikolova, V.; Koo, C.Y.; Ibrahim, S.A.; Wang, Z.; Spillmann, D.; Dreier, R.; Kelsch, R.; Fischgräbe, J.; Smollich, M.; Rossi, L.H.; et al. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis 2009, 30, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Götte, M.; Kersting, C.; Ruggiero, M.; Tio, J.; Tulusan, A.H.; Kiesel, L.; Wülfing, P. Predictive value of syndecan-1 expression for the response to neoadjuvant chemotherapy of primary breast cancer. Anticancer Res. 2006, 26, 621–627. [Google Scholar]
- Shimada, K.; Nakamura, M.; Velasco, M.; Tanaka, M.; Ouji, Y.; Konishi, N. Syndecan-1, a new target molecule involved in progression of androgen-independent prostate cancer. Cancer Sci. 2009, 100, 1248–1254. [Google Scholar] [CrossRef]
- Roy, A.; Femel, J.; Huijbers, E.J.M.; Spillmann, D.; Larsson, E.; Ringvall, M.; Olsson, A.-K.; Åbrink, M. Targeting Serglycin Prevents Metastasis in Murine Mammary Carcinoma. PLoS ONE 2016, 11, e0156151. [Google Scholar] [CrossRef] [PubMed]
- Korpetinou, A.; Skandalis, S.S.; Moustakas, A.; Happonen, K.E.; Tveit, H.; Prydz, K.; Labropoulou, V.T.; Giannopoulou, E.; Kalofonos, H.P.; Blom, A.M.; et al. Serglycin Is Implicated in the Promotion of Aggressive Phenotype of Breast Cancer Cells. PLoS ONE 2013, 8, e78157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbiotti, M.A.; Buraschi, S.; Kapoor, A.; Iozzo, R.V. Proteoglycan signaling in tumor angiogenesis and endothelial cell autophagy. Semin. Cancer Biol. 2020, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Sasaki, T.; Gohring, W.; Yamada, Y.; Timpl, R. The C-terminal domain V of perlecan promotes beta1 integrin-mediated cell adhesion, binds heparin, nidogen and fibulin-2 and can be modified by glycosaminoglycans. Eur. J. Biochem. 1997, 250, 39–46. [Google Scholar] [CrossRef]
- Aviezer, D.; Hecht, D.; Safran, M.; Eisinger, M.; David, G.; Yayon, A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 1994, 79, 1005–1013. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Whitelock, J.M.; Iozzo, R.V. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. J. Int. Soc. Matrix Biol. 2009, 28, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Zoeller, J.J.; Nyström, A. Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef]
- Kapoor, A.; Chen, C.G.; Iozzo, R.V. Endorepellin evokes an angiostatic stress signaling cascade in endothelial cells. J. Biol. Chem. 2020, 295, 6344–6356. [Google Scholar] [CrossRef] [Green Version]
- Cailhier, J.-F.; Sirois, I.; Laplante, P.; Lepage, S.; Raymond, M.-A.; Brassard, N.; Prat, A.; Iozzo, R.V.; Pshezhetsky, A.V.; Hébert, M.-J. Caspase-3 Activation Triggers Extracellular Cathepsin L Release and Endorepellin Proteolysis. J. Biol. Chem. 2008, 283, 27220–27229. [Google Scholar] [CrossRef] [Green Version]
- Mongiat, M.; Sweeney, S.M.; Antonio, J.D.S.; Fu, J.; Iozzo, R.V. Endorepellin, a Novel Inhibitor of Angiogenesis Derived from the C Terminus of Perlecan. J. Biol. Chem. 2002, 278, 4238–4249. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Pal, N.; Concannon, M.; Paul, M.; Doran, M.; Poluzzi, C.; Sekiguchi, K.; Whitelock, J.M.; Neill, T.; Iozzo, R.V. Endorepellin, the angiostatic module of perlecan, interacts with both the alpha2beta1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2): A dual receptor antagonism. J. Biol. Chem. 2011, 286, 25947–25962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, H.R. Syndecans in the diagnosis and prognosis of prostate cancer. Rev. Médica Chile 2010, 138, 95–101. [Google Scholar] [PubMed]
- Maeda, T.; Desouky, J.; Friedl, A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene 2005, 25, 1408–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.; Ramus, M.D.; Kirkwood, C.T.; Sperry, E.E.; Chu, P.-H.; Kao, W.W.; Albig, A.R. Lumican Exhibits Anti-Angiogenic Activity in a Context Specific Manner. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2013, 6, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Turley, R.S.; Finger, E.C.; Hempel, N.; How, T.; Fields, T.A.; Blobe, G.C. The Type III Transforming Growth Factor-β Receptor as a Novel Tumor Suppressor Gene in Prostate Cancer. Cancer Res. 2007, 67, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Ajiboye, S.; Sissung, T.M.; Sharifi, N.; Figg, W.D. More than an accessory: Implications of type III transforming growth factor-beta receptor loss in prostate cancer. BJU Int. 2010, 105, 913–916. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Wang, L.; López-Casillas, F.; Mendoza, V.; Yeh, I.-T.; Sun, L.-Z. Systemic administration of a soluble betaglycan suppresses tumor growth, angiogenesis, and matrix metalloproteinase-9 expression in a human xenograft model of prostate cancer. Prostate 2004, 63, 81–90. [Google Scholar] [CrossRef]
- Roblek, M.; Strutzmann, E.; Zankl, C.; Adage, T.; Heikenwalder, M.; Atlić, A.; Weis, R.; Kungl, A.; Borsig, L. Targeting of CCL2-CCR2-Glycosaminoglycan Axis Using a CCL2 Decoy Protein Attenuates Metastasis through Inhibition of Tumor Cell Seeding. Neoplasia 2016, 18, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Soucek, P. Xenobiotics, 1st ed.; Springer: Berlin, Germany, 2011; pp. 343–345. [Google Scholar]
- Rouissi, K.; Bechr, H.; Slah, O.; Amel, B.E. Genomics views: Xenobiotic metabolizing enzymes and cancer risks. Arab J. Oncol. 2011, 4, 40–46. [Google Scholar]
- Casey, S.C.; Vaccari, M.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Barcellos-Hoff, M.H.; Brown, D.G.; Chapellier, M.; Christopher, J.; Curran, C.S.; et al. The effect of environmental chemicals on the tumor microenvironment. Carcinogenesis 2015, 36, S160–S183. [Google Scholar] [CrossRef]
- Caruntu, C.; Mirica, A.; Roşca, A.; Mirica, R.; Caruntu, A.; Tampa, M.; Matei, C.; Constantin, C.; Neagu, M.; Badarau, A.; et al. The Role of Estrogens and Estrogen Receptors in Melanoma Development and Progression. Acta Endocrinol. Buchar. 2016, 12, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Peremiquel-Trillas, P.; Benavente, Y.; Martín-Bustamante, M.; Casabonne, D.; Perez-Gomez, B.; Gómez-Acebo, I.; Oliete-Canela, A.; Diéguez-Rodríguez, M.; Tusquets, I.; Amiano, P.; et al. Alkylphenolic compounds and risk of breast and prostate cancer in the MCC-Spain study. Environ. Int. 2019, 122, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Gatenby, R.A. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007, 26, 311–317. [Google Scholar] [CrossRef]
- Laconi, E. The evolving concept of tumor microenvironments. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 738–744. [Google Scholar] [CrossRef]
- Marongiu, F.; Doratiotto, S.; Sini, M.; Serra, M.P.; Laconi, E. Cancer as a disease of tissue pattern formation. Prog. Histochem. Cytochem. 2012, 47, 175–207. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Maté, I.; A Madrid, J.; Fuente, M. Chronobiology of the Neuroimmunoendocrine System and Aging. Curr. Pharm. Des. 2014, 20, 4642–4655. [Google Scholar] [CrossRef]
- Murphy, W.J.; Rui, H.; Longo, D.L. Effects of growth hormone and prolactin immune development and function. Life Sci. 1995, 57, 1–14. [Google Scholar] [CrossRef]
- Kopras, E.; Potluri, V.; Bermudez, M.-L.; Williams, K.; Belcher, S.; Kasper, S. Actions of endocrine-disrupting chemicals on stem/progenitor cells during development and disease. Endocr.-Relat. Cancer 2014, 21, T1–T12. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, D.; Margina, D.; Tsarouhas, K.; Tekos, F.; Stan, M.; Nikitovic, D.; Kouretas, D.; Spandidos, D.A.; Tsatsakis, A. Obesity a risk factor for increased COVID19 prevalence, severity and lethality (Review). Mol. Med. Rep. 2020, 22, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, A.; Szybiak, A.K.; Rachoń, D.; Serkies, K. Endocrine disrupting chemicals as the potential risk factor for estrogen-dependent cancers. Pol. Arch. Med. Wewn. 2016, 126, 562–570. [Google Scholar] [CrossRef]
- Vassilopoulou, L.; Psycharakis, C.; Petrakis, D.; Tsiaoussis, J.; Tsatsakis, A.M. Obesity, Persistent Organic Pollutants and Related Health Problems. Adv. Exp. Med. Biol. 2017, 960, 81–110. [Google Scholar]
- Neagu, M.; Constantin, C.; Popescu, I.D.; Zipeto, D.; Tzanakakis, G.; Nikitovic, D.; Fenga, C.; Stratakis, C.A.; Spandidos, D.A.; Tsatsakis, A. Inflammation and Metabolism in Cancer Cell—Mitochondria Key Player. Front. Oncol. 2019, 9, 348. [Google Scholar] [CrossRef] [Green Version]
- Petrakis, D.; Vassilopoulou, L.; Mamoulakis, C.; Psycharakis, C.; Anifantaki, A.; Sifakis, S.; Docea, A.O.; Tsiaoussis, J.; Makrigiannakis, A.; Tsatsakis, A. Endocrine Disruptors Leading to Obesity and Related Diseases. Int. J. Environ. Res. Public Health 2017, 14, 1282. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Nikitovic, D.; Berdiaki, A.; Spyridaki, I.; Krasanakis, T.; Tsatsakis, A.; Tzanakakis, G.N. Proteoglycans—Biomarkers and Targets in Cancer Therapy. Front. Endocrinol. 2018, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Xu, T.; Xu, X.; Cui, Y.; Xing, X. Biglycan promotes the chemotherapy resistance of colon cancer by activating NF-kappaB signal transduction. Mol. Cell. Biochem. 2018, 449, 285–294. [Google Scholar] [CrossRef]
- Schaefer, L.; Bábelová, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Brodbeck, R.; Bábelová, A.; Gretz, N.; Spieker, T.; Zeng-Brouwers, J.; Pfeilschifter, J.; Young, M.F.; Schaefer, R.M.; Schaefer, L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Investig. 2010, 120, 4251–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merline, R.; Moreth, K.; Beckmann, J.; Nastase, M.V.; Zeng-Brouwers, J.; Tralhão, J.G.; LeMarchand, P.; Pfeilschifter, J.; Schaefer, R.M.; Iozzo, R.V.; et al. Signaling by the Matrix Proteoglycan Decorin Controls Inflammation and Cancer Through PDCD4 and MicroRNA-21. Sci. Signal. 2011, 4, ra75. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, G.-L.; Jia, W.-D.; Ma, J.-L.; Li, J.-S.; Ge, Y.-S.; Ren, W.-H.; Yu, J.-H.; Liu, W.-B. Ligation of TLR2 by Versican: A Link between Inflammation and Metastasis. Arch. Med. Res. 2009, 40, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Diaby, V.; Tawk, R.; Sanogo, V.; Xiao, H.; Montero, A.J. A review of systematic reviews of the cost-effectiveness of hormone therapy, chemotherapy, and targeted therapy for breast cancer. Breast Cancer Res. Treat. 2015, 151, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Mazzoccoli, G.; Vendemiale, G.; De Cata, A.; Carughi, S.; Tarquini, R. Altered time structure of neuro-endocrine-immune system function in lung cancer patients. BMC Cancer 2010, 10, 314. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Ciavattini, A.; Petraglia, F.; Castellucci, M.; Ciarmela, P. Extracellular matrix in uterine leiomyoma pathogenesis: A potential target for future therapeutics. Hum. Reprod. Updat. 2017, 24, 59–85. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O.; Naba, A. Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2011, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Hubka, K.; Carson, D.D.; Harrington, D.A.; Farach-Carson, M.C.; Hubka, K.; Harrington, D.A.; Farach-Carson, M.C. Perlecan domain I gradients establish stable biomimetic heparin binding growth factor gradients for cell migration in hydrogels. Acta Biomater. 2019, 97, 385–398. [Google Scholar] [CrossRef]
- Steiner, M.L.; Theodoro, T.R.; Garcia, S.G.; Mader, A.; Pompei, L.M.; Pinhal, M.; Fernandes, C.E. Is the Expression of the Components of the Carotid Matrix of Rats Influenced by Estrogen, Progestin and Tibolone? Rev. Bras. Ginecol. Obstet. 2019, 41, 449–453. [Google Scholar] [CrossRef]
- Gonzalez, G.; Lage, A. Cancer vaccines for hormone/growth factor immune deprivation: A feasible approach for cancer treatment. Curr. Cancer Drug Targets 2007, 7, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive effects of androgens on the immune system. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Roved, J.; Westerdahl, H.; Hasselquist, D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm. Behav. 2017, 88, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Chang, J.J.; Chan, E.S.; Pollard, R.B.; Sidhu, H.K.; Kulkarni, S.; Wen, T.F.; Lindsay, R.J.; Orellana, L.; Mildvan, D.; et al. Sex differences in the Toll-like receptor–mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 2009, 15, 955–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesbeck, M.; Ziegler, S.; Laffont, S.; Smith, N.; Chauveau, L.; Tomezsko, P.; Sharei, A.; Kourjian, G.; Porichis, F.; Hart, M.; et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-α Production in Women. J. Immunol. 2015, 195, 5327–5336. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.; Ahmed, S.A. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Ellem, S.J.; Schmitt, J.F.; Pedersen, J.S.; Frydenberg, M.; Risbridger, G.P. Local Aromatase Expression in Human Prostate Is Altered in Malignancy. J. Clin. Endocrinol. Metab. 2004, 89, 2434–2441. [Google Scholar] [CrossRef] [Green Version]
- Bosland, M.C.; Mahmoud, A.M. Hormones and prostate carcinogenesis: Androgens and estrogens. J. Carcinog. 2011, 10, 33. [Google Scholar] [CrossRef]
- Crespi, E.; Bottai, G.; Santarpia, L. Role of inflammation in obesity-related breast cancer. Curr. Opin. Pharmacol. 2016, 31, 114–122. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, S.; Venditti, C.A.; Pei, X.-Y.; Nguyen, T.K.; Dent, P.; Grant, S. Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood 2008, 112, 793–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovats, S.; Carreras, E.; Agrawal, H. Sex Steroid Receptors in Immune Cells. In Sex Hormones and Immunity to Infection; Klein, S., Roberts, C., Eds.; Springer: Berlin, Germany, 2010. [Google Scholar]
- Kissick, H.T.; Sanda, M.G.; Dunn, L.K.; Pellegrini, K.L.; On, S.T.; Noel, J.K.; Arredouani, M.S. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 9887–9892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, S.T.; Lin, D.W.; Mostaghel, E.A.; Marck, B.T.; Wright, J.L.; Wu, J.; Amory, J.K.; Nelson, P.S.; Matsumoto, A.M. Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: A randomized-controlled trial. J. Clin. Endocrinol. Metab. 2010, 96, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Need, E.F.; Atashgaran, V.; Ingman, W.V.; Dasari, P. Hormonal Regulation of the Immune Microenvironment in the Mammary Gland. J. Mammary Gland. Biol. Neoplasia 2014, 19, 229–239. [Google Scholar] [CrossRef]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Stute, P.; Sielker, S.; Wood, C.E.; Register, T.C.; Lees, C.J.; Dewi, F.N.; Williams, J.K.; Wagner, J.D.; Stefenelli, U.; Cline, J.M. Life stage differences in mammary gland gene expression profile in non-human primates. Breast Cancer Res. Treat. 2011, 133, 617–634. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, A.; Allavena, P.; Santoro, G.; Fumarulo, R.; Corsi, M.M.; Mantovani, A. Molecular pathways in cancer-related inflammation. Biochem. Medica 2011, 21, 264–275. [Google Scholar] [CrossRef]
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M. Estrogen Receptor-Alpha Promotes Alternative Macrophage Activation during Cutaneous Repair. J. Investig. Dermatol. 2014, 134, 2447–2457. [Google Scholar] [CrossRef] [Green Version]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in Inflammatory Breast Carcinoma Incidence and Survival: The Surveillance, Epidemiology, and End Results Program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Gotte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.E.; Gadalla, R.; Hassan, H.; Afifi, A.; Götte, M.; El-Shinawi, M.; Mohamed, M.M.; Ibrahim, S.A. The immunomodulatory role of tumor Syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS ONE 2019, 14, e0217550. [Google Scholar] [CrossRef] [Green Version]
- Sue, M.; Higashi, N.; Shida, H.; Kogane, Y.; Nishimura, Y.; Adachi, H.; Kolaczkowska, E.; Kepka, M.; Nakajima, M.; Irimura, T. An iminosugar-based heparanase inhibitor heparastatin (SF4) suppresses infiltration of neutrophils and monocytes into inflamed dorsal air pouches. Int. Immunopharmacol. 2016, 35, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, G.; Saarinen, N.; Abrahamsson, A.; Dabrosin, C. Tamoxifen, Flaxseed, and the Lignan Enterolactone Increase Stroma- and Cancer Cell-Derived IL-1Ra and Decrease Tumor Angiogenesis in Estrogen-Dependent Breast Cancer. Cancer Res. 2010, 71, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Simões, B.M.; Howell, S.J.; Farnie, G.; Clarke, R. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. BCR 2013, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Ancuceanu, R.; Neagu, M. Immune based therapy for melanoma. Indian J. Med. Res. 2016, 143, 135–144. [Google Scholar] [CrossRef]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, S.; Schuetz, F.; Sohn, C.; Beckhove, P.; Domschke, C. Adoptive immunotherapy of metastatic breast cancer: Present and future. Cancer Metastasis Rev. 2013, 33, 309–320. [Google Scholar] [CrossRef]
- Boibessot, C.; Toren, P. Sex steroids in the tumor microenvironment and prostate cancer progression. Endocr.-Relat. Cancer 2018, 25, R179–R196. [Google Scholar] [CrossRef]
- Ylitalo, E.B.; Thysell, E.; Jernberg, E.; Lundholm, M.; Crnalic, S.; Egevad, L.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Subgroups of Castration-resistant Prostate Cancer Bone Metastases Defined Through an Inverse Relationship Between Androgen Receptor Activity and Immune Response. Eur. Urol. 2017, 71, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strömvall, K.; Sundkvist, K.; Ljungberg, B.; Bergström, S.H.; Bergh, A. Reduced number of CD169+macrophages in pre-metastatic regional lymph nodes is associated with subsequent metastatic disease in an animal model and with poor outcome in prostate cancer patients. Prostate 2017, 77, 1468–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrossay, D.; Thelen, M. Immune response: Steroids drive dendritic cells. Nat. Immunol. 2013, 14, 424–426. [Google Scholar] [CrossRef]
- Bouman, A.; Heineman, M.J.; Faas, M.M. Sex hormones and the immune response in humans. Hum. Reprod. Updat. 2005, 11, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, M.P.; Meydani, M.; Lichtenstein, A.H.; Schaefer, E.J.; Dillard, A.; Lamon-Fava, S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J. Endocrinol. 2010, 206, 217–224. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, P.; Milano, S.; Barbera, C.; Di Bella, G.; La Rosa, M.; Ferlazzo, V.; Farruggio, R.; Miceli, D.M.; Miele, M.; Castagnetta, L.; et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann. N. Y. Acad. Sci. 1999, 876, 426–429. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone Reduces Macrophage Expression in the Mouse of Toll-Like Receptor 4, a Trigger for Inflammation and Innate Immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef]
- Pioli, P.A.; Jensen, A.L.; Weaver, L.K.; Amiel, E.; Shen, Z.; Shen, L.; Wira, C.R.; Guyre, P.M. Estradiol attenuates lipopolysaccharide-induced CXC chemokine ligand 8 production by human peripheral blood monocytes. J. Immunol. 2007, 179, 6284–6290. [Google Scholar] [CrossRef] [Green Version]
- Hood, S.P.; Foulds, G.A.; Imrie, H.; Reeder, S.; McArdle, S.E.B.; Khan, M.; Pockley, A.G. Phenotype and Function of Activated Natural Killer Cells From Patients With Prostate Cancer: Patient-Dependent Responses to Priming and IL-2 Activation. Front. Immunol. 2019, 9, 3169. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sánchez-López, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Li, P.; Peng, F.; Zhang, M.; Zhang, Y.Y.; Liang, H.; Zhao, W.; Qi, L.; Wang, H.; Wang, C.; et al. Autophagy-related prognostic signature for breast cancer. Mol. Carcinog. 2015, 55, 292–299. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.-M.; Bourachot, B.; Zago, G.; Bièche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2015, 10, 2122–2142. [Google Scholar] [CrossRef]
- Park, J.-H.; Kim, K.P.; Ko, J.-J.; Park, K. PI3K/Akt/mTOR activation by suppression of ELK3 mediates chemosensitivity of MDA-MB-231 cells to doxorubicin by inhibiting autophagy. Biochem. Biophys. Res. Commun. 2016, 477, 277–282. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. AMPK and Cancer. Exp. Suppl. 2016, 107, 203–226. [Google Scholar]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Müller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, S.; Dufies, M.; Ndiaye, P.D.; Viotti, J.; Borchiellini, D.; Parola, J.; Vial, V.; Cormerais, Y.; Ohanna, M.; Imbert, V.; et al. Resistance to lysosomotropic drugs used to treat kidney and breast cancers involves autophagy and inflammation and converges in inducing CXCL5. Theranostics 2019, 9, 1181–1199. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Masuda, N.; Kamigaki, S.; Morimoto, T.; Saji, S.; Imoto, S.; Sasano, H.; Toi, M. Differential Involvement of Autophagy and Apoptosis in Response to Chemoendocrine and Endocrine Therapy in Breast Cancer: JBCRG-07TR. Int. J. Mol. Sci. 2019, 20, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Saji, S.; Sugimoto, M.; Masuda, N.; Kuroi, K.; Sato, N.; Takei, H.; Yamamoto, Y.; Ohno, S.; Yamashita, H.; et al. Clinical significance of the expression of autophagy-associated marker, beclin 1, in breast cancer patients who received neoadjuvant endocrine therapy. BMC Cancer 2016, 16, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoureux, F.; Zoubeidi, A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy 2013, 9, 1119–1120. [Google Scholar] [CrossRef] [Green Version]
- Damodaran, S.; Lang, J.M.; Jarrard, D.F. Targeting Metastatic Hormone Sensitive Prostate Cancer: Chemohormonal Therapy and New Combinatorial Approaches. J. Urol. 2019, 201, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Neill, T.; Owens, R.T.; Schaefer, L.; Iozzo, R.V. Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 34, 46–54. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Seifert, E.L.; Rodeck, U.; Hoek, J.B.; Iozzo, R.V. Metabolic reprogramming of murine cardiomyocytes during autophagy requires the extracellular nutrient sensor decorin. J. Biol. Chem. 2018, 293, 16940–16950. [Google Scholar] [CrossRef] [Green Version]
- Neill, T.; Torres, A.; Buraschi, S.; Owens, R.T.; Hoek, J.B.; Baffa, R.; Iozzo, R.V. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) and mitostatin. J. Biol. Chem. 2014, 289, 4952–4968. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Xu, W.; Neill, T.; Hu, Z.; Wang, C.-H.; Xiao, X.; Stock, S.R.; Guise, T.; Yun, C.-O.; Brendler, C.B.; et al. Systemic Delivery of an Oncolytic Adenovirus Expressing Decorin for the Treatment of Breast Cancer Bone Metastases. Hum. Gene Ther. 2015, 26, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Zhang, C.G.; Gong, M.T.; Zhang, M.; Wang, L.; Ding, W. Decorin-mediated inhibition of the migration of U87MG glioma cells involves activation of autophagy and suppression of TGF-beta signaling. FEBS Open Bio 2016, 6, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Yang, B.B.; Yang, B.L.; Deng, Z.; Fang, L.; Shan, S.W.; Jeyapalan, Z.; Zhang, Y.; Seth, A.; Yee, A.J. Versican G3 domain modulates breast cancer cell apoptosis: A mechanism for breast cancer cell response to chemotherapy and EGFR therapy. PLoS ONE 2011, 6, e26396. [Google Scholar] [CrossRef] [PubMed]
- Sooro, M.A.; Zhang, N.; Zhang, P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 2018, 143, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Seidler, D.G.; Goldoni, S.; Agnew, C.; Cardi, C.; Thakur, M.L.; Owens, R.T.; McQuillan, D.J.; Iozzo, R.V. Decorin Protein Core Inhibitsin VivoCancer Growth and Metabolism by Hindering Epidermal Growth Factor Receptor Function and Triggering Apoptosis via Caspase-3 Activation. J. Biol. Chem. 2006, 281, 26408–26418. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef]
- Yoon, A.-R.; Hong, J.; Yun, C.-O. Adenovirus-mediated decorin expression induces cancer cell death through activation of p53 and mitochondrial apoptosis. Oncotarget 2017, 8, 76666–76685. [Google Scholar] [CrossRef] [Green Version]
- Roedig, H.; Nastase, M.V.; Frey, H.; Moreth, K.; Zeng-Brouwers, J.; Poluzzi, C.; Hsieh, L.T.-H.; Brandts, C.; Fulda, S.; Wygrecka, M.; et al. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 77, 4–22. [Google Scholar] [CrossRef]
- Socovich, A.M.; Naba, A. The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef]
- Tóth, I.Y.; Illes, E.; Szekeres, M.; Zupkó, I.; Turcu, R.; Tombácz, E. Chondroitin-Sulfate-A-Coated Magnetite Nanoparticles: Synthesis, Characterization and Testing to Predict Their Colloidal Behavior in Biological Milieu. Int. J. Mol. Sci. 2019, 20, 4096. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulou, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. The biological role of chondroitin sulfate in cancer and chondroitin-based anticancer agents. Vivo 2008, 22, 385–389. [Google Scholar]
- Bagari, R.; Bansal, D.; Gulbake, A.; Jain, A.; Soni, V.; Jain, S.K. Chondroitin sulfate functionalized liposomes for solid tumor targeting. J. Drug Target. 2010, 19, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Park, S.-J.; Na, K. Potential of self-organizing nanogel with acetylated chondroitin sulfate as an anti-cancer drug carrier. Colloids Surf. B Biointerfaces 2010, 79, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J. 2003 Claude, S. Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity. J. Med. Chem. 2003, 46, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Moon, H.T.; Park, G.-E.; Jeon, O.C.; Byun, Y.; Lee, Y.-K. Preparation of Sodium Deoxycholate (DOC) Conjugated Heparin Derivatives for Inhibition of Angiogenesis and Cancer Cell Growth. Bioconjug. Chem. 2008, 19, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- She, W.; Li, N.; Luo, K.; Guo, C.; Wang, G.; Geng, Y.; Gu, Z. Dendronized heparin–doxorubicin conjugate based nanoparticle as pH-responsive drug delivery system for cancer therapy. Biomaterials 2013, 34, 2252–2264. [Google Scholar] [CrossRef]
- Lanzi, C.; Zaffaroni, N.; Cassinelli, G.; Lanzi, N.Z.A.G.C.C. Targeting Heparan Sulfate Proteoglycans and their Modifying Enzymes to Enhance Anticancer Chemotherapy Efficacy and Overcome Drug Resistance. Curr. Med. Chem. 2017, 24, 2860–2886. [Google Scholar] [CrossRef]
- Coombe, D.R.; Gandhi, N.S. Heparanase: A Challenging Cancer Drug Target. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Lerner, I.; Baraz, L.; Pikarsky, E.; Meirovitz, A.; Edovitsky, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Function of Heparanase in Prostate Tumorigenesis: Potential for Therapy. Clin. Cancer Res. 2008, 14, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Onyeisi, J.O.S.; Filho, P.C.D.A.P.; Lopes, S.D.A.; Nader, H.B.; Lopes, C.C. Heparan sulfate proteoglycans as trastuzumab targets in anoikis-resistant endothelial cells. J. Cell. Biochem. 2019, 120, 13826–13840. [Google Scholar] [CrossRef]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides—Challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzanakakis, G.; Giatagana, E.-M.; Kuskov, A.; Berdiaki, A.; Tsatsakis, A.; Neagu, M.; Nikitovic, D. Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors. Cancers 2020, 12, 2401. https://doi.org/10.3390/cancers12092401
Tzanakakis G, Giatagana E-M, Kuskov A, Berdiaki A, Tsatsakis A, Neagu M, Nikitovic D. Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors. Cancers. 2020; 12(9):2401. https://doi.org/10.3390/cancers12092401
Chicago/Turabian StyleTzanakakis, George, Eirini-Maria Giatagana, Andrey Kuskov, Aikaterini Berdiaki, Aristidis Tsatsakis, Monica Neagu, and Dragana Nikitovic. 2020. "Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors" Cancers 12, no. 9: 2401. https://doi.org/10.3390/cancers12092401
APA StyleTzanakakis, G., Giatagana, E. -M., Kuskov, A., Berdiaki, A., Tsatsakis, A., Neagu, M., & Nikitovic, D. (2020). Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors. Cancers, 12(9), 2401. https://doi.org/10.3390/cancers12092401