Development of a Cationic Amphiphilic Helical Peptidomimetic (B18L) As A Novel Anti-Cancer Drug Lead



Abstract
:Simple Summary
Abstract
1. Introduction
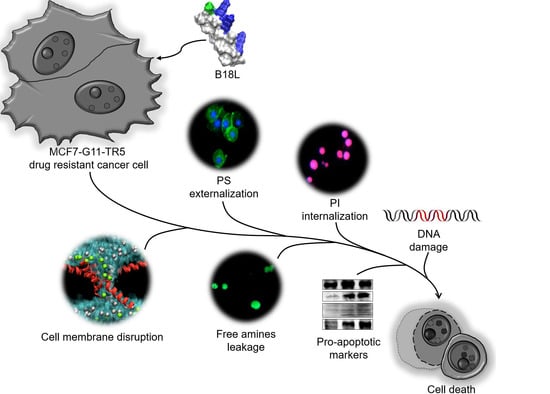
2. Results
2.1. Design of B18-Based Peptidomimetics
2.2. Design of B18K—Role of Polar Positive Amino Acids Alone
2.3. Design of B18KL and B18KA—Role of Hydrophobic Amino Acids
2.4. Design of B18L as a Compound Lead—Role of Branched-Chain Amino Acids (BCAAs)
2.5. B18L As a Compound Lead—Activity Toward Drug-Resistant Breast Cancer (DRBC) Cells
2.6. Structure of B18L
2.7. B18L Binds BST-2 Protein
2.8. B18L Is Less Toxic to Normal Human Cells
2.9. B18L Causes Early DRBC Apoptotic Cell Death and Triggers Intracellular Signal Transduction
2.10. Structural Dynamics of B18L on the Cell Membrane as Assessed By Molecular Dynamics (MD) Simulations
2.11. B18L Impairs Cancer Cell Membrane Integrity
2.12. B18L Potentiates Phosphatidylserine (PS) Externalization in the Plasma Membranes
2.13. B18L Activates Integrin Alpha 5 and Beta 1 Expression and Induces Sustained Inhibition of Src and Erk1/2 Activation
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Chemical Reagents
4.3. Cell Lines
4.4. Peptide Synthesis
4.5. CD Analysis
4.6. Viability Assay
4.7. Hemolysis Assay
4.8. Real-Time Quantitative PCR (RT-qPCR)
4.9. Western Blot
4.10. Molecular Dynamics (MD) Simulation
4.11. Analysis of Cell Morphology by Live-Imaging Microscopy
4.12. Annexin V and PI Staining Assay
4.13. UV Spectroscopy Assay
4.14. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eggermont, A.M.G.; Kroemer, G.; Zitvogel, L. Immunotherapy and the concept of a clinical cure. Eur. J. Cancer 2013, 49, 2965–2967. [Google Scholar] [CrossRef] [PubMed]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. B49, a BST-2-based peptide, inhibits adhesion and growth of breast cancer cells. Sci. Rep. 2018, 8, 4305. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2008, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.H.; Mehta, H.V.; Maric, M.; Roller, R.J.; Okeoma, C.M. Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology 2012, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Wang, J.-J.; Qi, M.; Yoon, J.-J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.H.; Okeoma, C.M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated BST-2/tetherin regulation. Cell Signal 2013, 25, 2752–2761. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. Cysteine-linked dimerization of BST-2 confers anoikis resistance to breast cancer cells by negating proapoptotic activities to promote tumor cell survival and growth. Cell Death Dis. 2017, 8, e2687. [Google Scholar] [CrossRef] [Green Version]
- Mahauad-Fernandez, W.D.; Borcherding, N.C.; Zhang, W.; Okeoma, C.M. Bone marrow stromal antigen 2 (BST-2) DNA is demethylated in breast tumors and breast cancer cells. PLoS ONE 2015, 10, e0123931. [Google Scholar] [CrossRef] [Green Version]
- Lyu, Y.; Mahauad-Fernandez, W.D.; Okeoma, C.M. Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1. Molecules 2020, 25, 1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Yumoto, K.; Yasuhara, K.; Nadres, E.T.; Kikuchi, Y.; Buttitta, L.; Taichman, R.S.; Kuroda, K. Anticancer polymers designed for killing dormant prostate cancer cells. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, L.S.; Marquette, A.; Schoup, M.; Fenard, D.; Galy, A.; Bechinger, B. Simultaneous analysis of secondary structure and light scattering from circular dichroism titrations: Application to vectofusin-1. Sci. Rep. 2016, 6, 39450. [Google Scholar] [CrossRef] [Green Version]
- Carretero, G.; Saraiva, G.K.; Cauz, A.C.; Rodrigues, M.A.; Kiyota, S.; Riske, K.A.; Dos Santos, A.A.; Pinatto-Botelho, M.F.; Bemquerer, M.P.; Gueirosfilho, F.; et al. Synthesis, biophysical and functional studies of two BP100 analogues modified by a hydrophobic chain and a cyclic peptide. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 1502–1516. [Google Scholar] [CrossRef]
- Zhang, M.-K.; Lyu, Y.; Zhu, X.; Wang, J.; Jin, Z.-Y.; Narsimhan, G. Enhanced solubility and antimicrobial activity of alamethicin in aqueous solution by complexation with γ-cyclodextrin. J. Funct. Foods 2018, 40, 700–706. [Google Scholar] [CrossRef]
- Metsämuuronen, S.; Mänttäri, M.; Nyström, M. Comparison of analysis methods for protein concentration and its use in UF fractionation of whey. Desalination 2011, 283, 156–164. [Google Scholar] [CrossRef]
- Deb, D.D.; Parimala, G.; Devi, S.S.; Chakraborty, T. Effect of thymol on peripheral blood mononuclear cell PBMC and acute promyelotic cancer cell line HL-60. Chem. Interact. 2011, 193, 97–106. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Armstrong, R.C.; Krebs, J.; Srinivasula, S.M.; Wang, L.; Bullrich, F.; Fritz, L.C.; Trapani, J.A.; Tomaselli, K.J.; Litwack, G.; et al. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad Sci. USA 1996, 93, 7464–7469. [Google Scholar] [CrossRef] [Green Version]
- Anguissola, S.; Köhler, B.; O’Byrne, R.; Düssmann, H.; Cannon, M.D.; Murray, F.E.; Concannon, C.G.; Rehm, M.; Kögel, D.; Prehn, J.H.M. Bid and calpains cooperate to trigger oxaliplatin-induced apoptosis of cervical carcinoma HeLa cells. Mol. Pharmacol. 2009, 76, 998–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.; Bennett, L.L. Resveratrol enhances the efficacy of sorafenib mediated apoptosis in human breast cancer MCF7 cells through ROS, cell cycle inhibition, caspase 3 and PARP cleavage. Biomed. Pharmacother. 2016, 84, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Cui, Y.-C.; Zhang, H.; Liu, X.-P.; Wu, A.-L.; Li, J.-J.; Tang, Y.; Zhang, N. Glutamine reduces the apoptosis of H9C2 cells treated with high-glucose and reperfusion through an oxidation-related mechanism. PLoS ONE 2015, 10, e0132402. [Google Scholar] [CrossRef] [PubMed]
- Diantini, A.; Subarnas, A.; Lestari, K.; Halimah, E.; Susilawati, Y.; Supriyatna; Julaeha, E.; Achmad, T.H.; Suradji, E.W.; Yamazaki, C.; et al. Kaempferol-3-O-rhamnoside isolated from the leaves of Schima wallichii Korth. inhibits MCF-7 breast cancer cell proliferation through activation of the caspase cascade pathway. Oncol. Lett. 2012, 3, 1069–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmschneider, M.B.; Ulmschneider, J.P.; Schiller, N.; Wallace, B.; Von Heijne, G.; White, S. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Farrotti, A.; Bocchinfuso, G.; Palleschi, A.; Rosato, N.; Salnikov, E.; Voievoda, N.; Bechinger, B.; Stella, L. Molecular dynamics methods to predict peptide locations in membranes: LAH4 as a stringent test case. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 581–592. [Google Scholar] [CrossRef]
- Lyu, Y.; Zhu, X.; Xiang, N.; Narsimhan, G. Molecular dynamics study of pore formation by melittin in a 1, 2-Dioleoyl-sn-glycero-3-phosphocholine and 1, 2-di (9 z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) mixed lipid bilayer. Ind. Eng. Chem. Res. 2015, 54, 10275–10283. [Google Scholar] [CrossRef]
- Lee, M.-S.; Kim, Y.B.; Lee, S.-Y.; Kim, J.-G.; Kim, S.-H.; Ye, S.-K.; Lee, J.W. Integrin signaling and cell spreading mediated by phorbol 12-myristate 13-acetate treatment. J. Cell. Biochem. 2006, 99, 88–95. [Google Scholar] [CrossRef]
- Ju, J.A.; Godet, I.; Ye, I.C.; Byun, J.; Jayatilaka, H.; Lee, S.J.; Xiang, L.; Samanta, D.; Lee, M.H.; Wu, P.-H.; et al. Hypoxia selectively enhances integrin alpha5beta1 receptor expression in breast cancer to promote metastasis. Mol. Cancer Res. 2017, 15, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Cao, Y.; Guan, Y.; Zheng, C. BST2 promotes cell proliferation, migration and induces NF-κB activation in gastric cancer. Biotechnol. Lett. 2018, 40, 1015–1027. [Google Scholar] [CrossRef]
- Mukai, S.; Oue, N.; Oshima, T.; Mukai, R.; Tatsumoto, Y.; Sakamoto, N.; Sentani, K.; Tanabe, K.; Egi, H.; Hinoi, T.; et al. Overexpression of transmembrane protein BST2 is associated with poor survival of patients with esophageal, gastric, or colorectal cancer. Ann. Surg. Oncol. 2017, 24, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.H.; Mahauad-Fernandez, W.D.; Madison, M.N.; Okeoma, C.M. BST-2/tetherin is overexpressed in mammary gland and tumor tissues in MMTV-induced mammary cancer. Virology 2013, 444, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with dual antimicrobial and anticancer activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.J.; Toombs, J.E.; Minna, J.D.; Brekken, R.A.; Udugamasooriya, D.G. Identification of lipid-phosphatidylserine (PS) as the target of unbiasedly selected cancer specific peptide-peptoid hybrid PPS1. Oncotarget 2016, 7, 30678–30690. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.-R.; Zhang, Q.; Tian, X.-B.; Cao, Y.-M.; Liu, Z.-Q.; Fan, R.; Ding, X.-F.; Zhu, Z.; Chen, L.; Luo, S.-Z. From a pro-apoptotic peptide to a lytic peptide: One single residue mutation. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 1914–1925. [Google Scholar] [CrossRef]
- Jamadi, R.H.; Khalili, S.; Mirzapour, T.; Yaghoubi, H.; Hashemi, Z.S.; Mard-Soltani, M.; Jafarisani, M. Anticancer activity of brevinin-2R peptide and its Two analogues against myelogenous leukemia cell line as natural treatments: An in vitro study. Int. J. Pept. Res. Ther. 2019, 26, 1013–1020. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Horst, B.A.; Shu, C.; Chau, L.; Tsujiuchi, T.; Bruce, J.N.; Canoll, P.; Greene, L.A.; Angelastro, J.M.; Siegelin, M.D. A synthetic cell-penetrating dominant-negative ATF5 peptide exerts anticancer activity against a broad spectrum of treatment-resistant cancers. Clin. Cancer Res. 2016, 22, 4698–4711. [Google Scholar] [CrossRef] [Green Version]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef]
- Van Engeland, M.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24, 131–139. [Google Scholar] [CrossRef]
- Dumont, E.; Reutelingsperger, C.; Smits, J.; Daemen, M.; Doevendans, P.; Wellens, H.; Hofstra, L. Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat. Med. 2001, 7, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, L.; Liem, I.H.; Dumont, A.E.; Boersma, H.H.; Van Heerde, W.L.; Doevendans, A.P.; Demuinck, E.; Wellens, H.; Kemerink, G.J.; Reutelingsperger, C.P.; et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet 2000, 356, 209–212. [Google Scholar] [CrossRef]
- Narula, J.; Acio, E.R.; Narula, N.; Samuels, L.E.; Fyfe, B.; Wood, D.; Fitzpatrick, J.M.; Raghunath, P.; Tomaszewski, J.E.; Kelly, C.; et al. Annexin-V imaging for noninvasive detection of cardiac allograft rejection. Nat. Med. 2001, 7, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, Y.; Yuan, H.; Li, X.; Cheng, A.; Lou, H.-X. Solamargine, a steroidal alkaloid glycoside, induces oncosis in human K562 leukemia and squamous cell carcinoma KB cells. Cancer Chemother. Pharmacol. 2010, 67, 813–821. [Google Scholar] [CrossRef]
- Lyu, Y.; Fitriyanti, M.; Narsimhan, G. Nucleation and growth of pores in 1, 2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC)/cholesterol bilayer by antimicrobial peptides melittin, its mutants and cecropin P1. Colloids Surf. B Biointerfaces 2019, 173, 121–127. [Google Scholar] [CrossRef]
- Wiedemann, C.; Bellstedt, P.; Görlach, M. CAPITO—A web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 2013, 29, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.C.; Nelson, C.E.; Yu, S.S.; Beavers, K.R.; Kim, A.J.; Li, H.; Nelson, H.M.; Giorgio, T.D.; Duvall, C.L. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J. Vis. Exp. 2013, 73, e50166. [Google Scholar] [CrossRef] [Green Version]
- Mahauad-Fernandez, W.D.; Jones, P.H.; Okeoma, C.M. Critical role for bone marrow stromal antigen 2 in acute Chikungunya virus infection. J. Gen. Virol. 2014, 95, 2450–2461. [Google Scholar] [CrossRef]
- Xiang, N.; Lyu, Y.; Zhu, X.; Narsimhan, G. Investigation of the interaction of amyloid beta peptide (11–42) oligomers with a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane using molecular dynamics simulation. Phys. Chem. Chem. Phys. 2018, 20, 6817–6829. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Lyu, Y.; Fitriyanti, M.; Hou, H.; Jin, Z.-Y.; Zhu, X.; Narsimhan, G. Understanding the antimicrobial activity of water soluble γ-cyclodextrin/alamethicin complex. Colloids Surf. B Biointerfaces 2018, 172, 451–458. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A.D.J. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Grap. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Caballero, B.; Trugo, L.C.; Finglas, P.M. Encyclopedia of Food Sciences and Nutrition; Elsevier Science Ltd. Academic Press: Cambrige, MA, USA, 2003. [Google Scholar]
- Tognarelli, D.; Tsukamoto, A.; Caldwell, J.; Caldwell, W. Rapid peptide separation by supercritical fluid chromatography. Bioanalysis 2010, 2, 5–7. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | # of Residues | Charge | H 1 (kcal/mol) | μH 2 (kcal/mol) | MW 3 (kDa) |
|---|---|---|---|---|---|---|
| B18 | GFQDVEAQAATCNHTVMA | 18 | –2 | 0.358 | 0.222 | 1.893 |
| B18K | GFQKVKAQAATCNHTVMA | 18 | 2 | 0.326 | 0.224 | 1.905 |
| B18KL | GFQKVKAQALTCLHTVMA | 18 | 2 | 0.531 | 0.315 | 1.946 |
| B18KA | GFQKAKAKALACLAKALA | 18 | 4 | 0.357 | 0.375 | 1.803 |
| B18L | GLGKALAKALACLAKALA | 18 | 3 | 0.513 | 0.523 | 1.683 |
| B18I | GIGKAIAKAIACIAKAIA | 18 | 3 | 0.541 | 0.547 | 1.683 |
| Cell Lines | Subtype | IC50 (µM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ER | PR | HER2 | B18K | B18KL | B18KA | B18L | B18I | ||
| MDA-MB-468 | Basal A | - | - | - | *>131.0 | >128.5 | 135.6 ± 9.3 | 12.4 ± 1.4 | 16.6 ± 1.5 |
| MDA-MB-231 | Claudin-Low | - | - | - | >131.0 | >128.5 | 83.7 ± 20.1 | 7.2 ± 0.8 | 18.6 ± 2.4 |
| ZR-75-1 | Luminal A/B | + | +/- | - | >131.0 | >128.5 | 58.9 ± 2.0 | 5.2 ± 1.0 | 10.1 ± 1.8 |
| T47D | Luminal A | + | +/- | - | >131.0 | >128.5 | 78.5 ± 20.9 | 17.5 ± 6.5 | >148.5 |
| SKBR3 | HER2 | - | - | + | >131.0 | >128.5 | 43.5 ± 11.5 | 7.2 ± 1.0 | 18.1 ± 4.7 |
| Cell lines | Subtype | IC50 (µM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ER | PR | HER2 | B18K | B18KL | B18KA | B18L | B18I | ||
| MCF7 | Drug-sensitive | + | + | - | *>131.0 | >128.5 | 61.8 ± 3.9 | 5.9 ± 0.5 | 16.0 ± 1.6 |
| MCF7-G11-TR1 | Drug-resistant (1 µM) | + | + | - | >131.0 | >128.5 | 61.8 ± 5.1 | 6.2 ± 0.8 | 10.8 ± 2.0 |
| MCF7-G11-TR5 | Drug-resistant (5 µM) | + | + | - | >131.0 | >128.5 | 56.4 ± 5.0 | 3.8 ± 0.3 | 16.1 ± 4.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, Y.; Kopcho, S.; Alvarez, F.A.; Okeoma, B.C.; Okeoma, C.M. Development of a Cationic Amphiphilic Helical Peptidomimetic (B18L) As A Novel Anti-Cancer Drug Lead. Cancers 2020, 12, 2448. https://doi.org/10.3390/cancers12092448
Lyu Y, Kopcho S, Alvarez FA, Okeoma BC, Okeoma CM. Development of a Cationic Amphiphilic Helical Peptidomimetic (B18L) As A Novel Anti-Cancer Drug Lead. Cancers. 2020; 12(9):2448. https://doi.org/10.3390/cancers12092448
Chicago/Turabian StyleLyu, Yuan, Steven Kopcho, Folnetti A. Alvarez, Bryson C. Okeoma, and Chioma M. Okeoma. 2020. "Development of a Cationic Amphiphilic Helical Peptidomimetic (B18L) As A Novel Anti-Cancer Drug Lead" Cancers 12, no. 9: 2448. https://doi.org/10.3390/cancers12092448
APA StyleLyu, Y., Kopcho, S., Alvarez, F. A., Okeoma, B. C., & Okeoma, C. M. (2020). Development of a Cationic Amphiphilic Helical Peptidomimetic (B18L) As A Novel Anti-Cancer Drug Lead. Cancers, 12(9), 2448. https://doi.org/10.3390/cancers12092448