Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
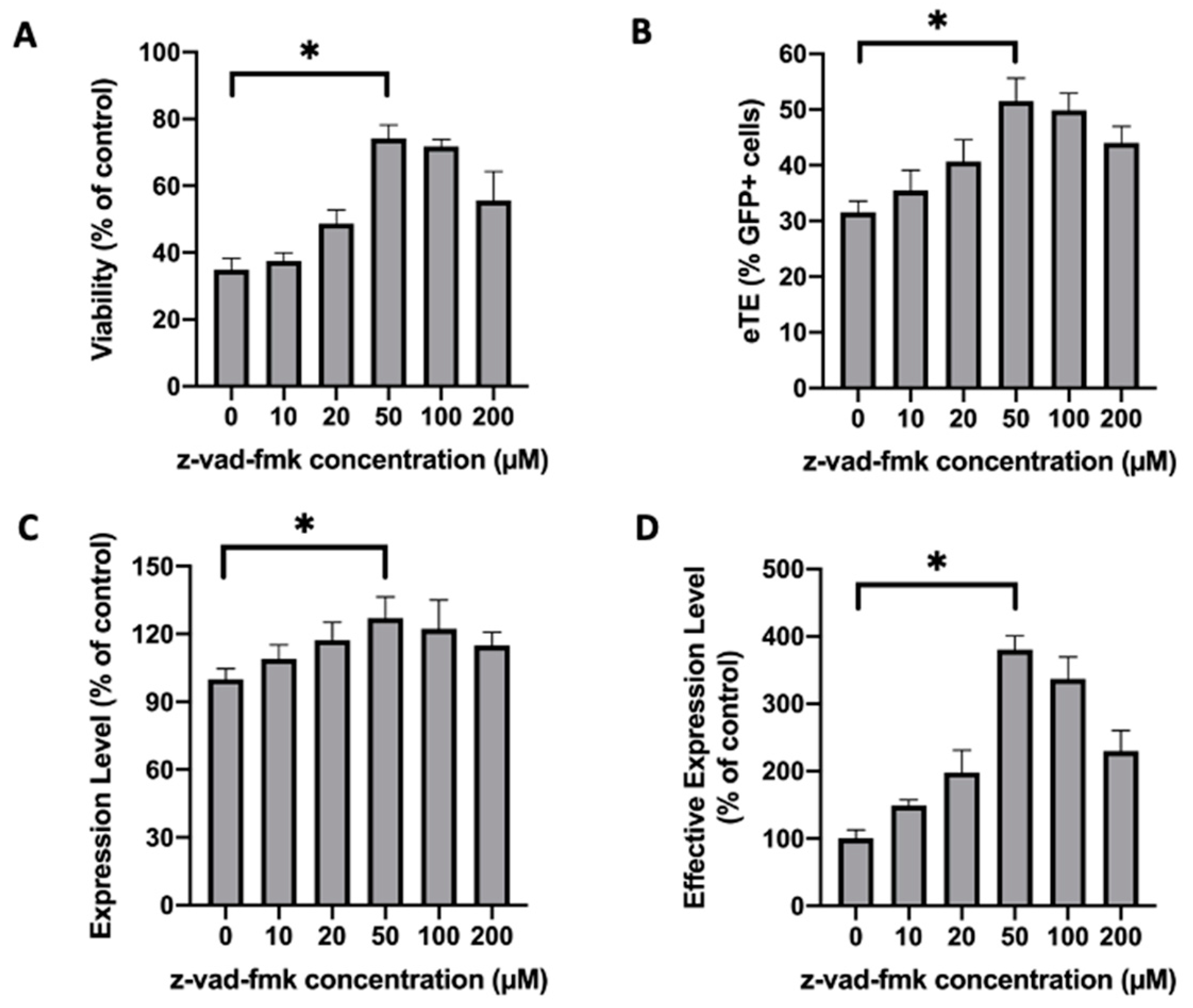
2.1. Treatment of Jurkat Cells with a Pan-Caspase Inhibitor Increases Cell Viability and Electrotransfer Efficiency
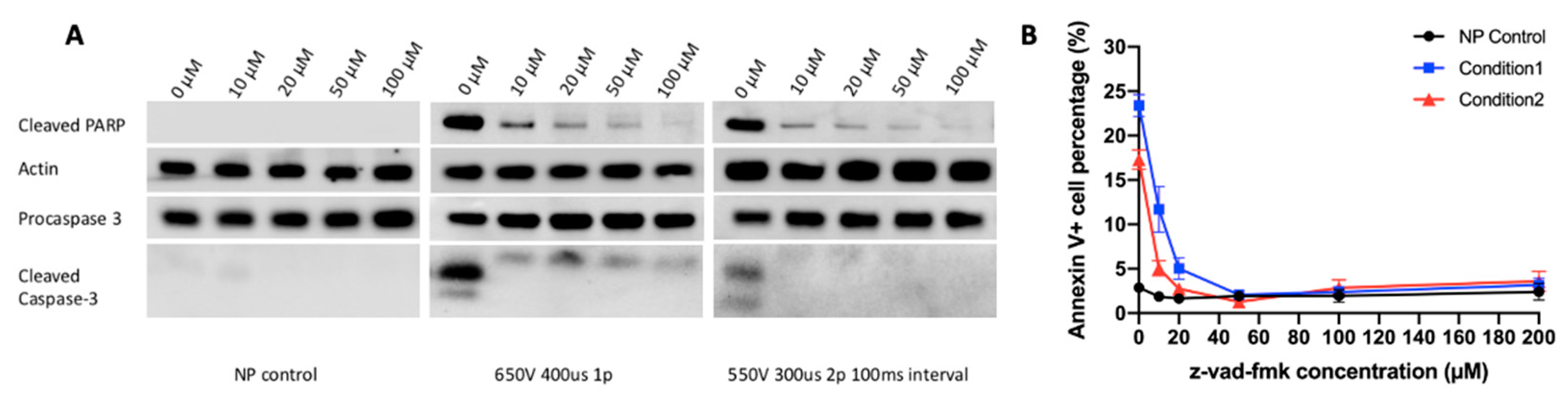
2.2. Caspase-Dependent Apoptosis Is an Important Mechanism of Electrotransfer-Induced Cell Death
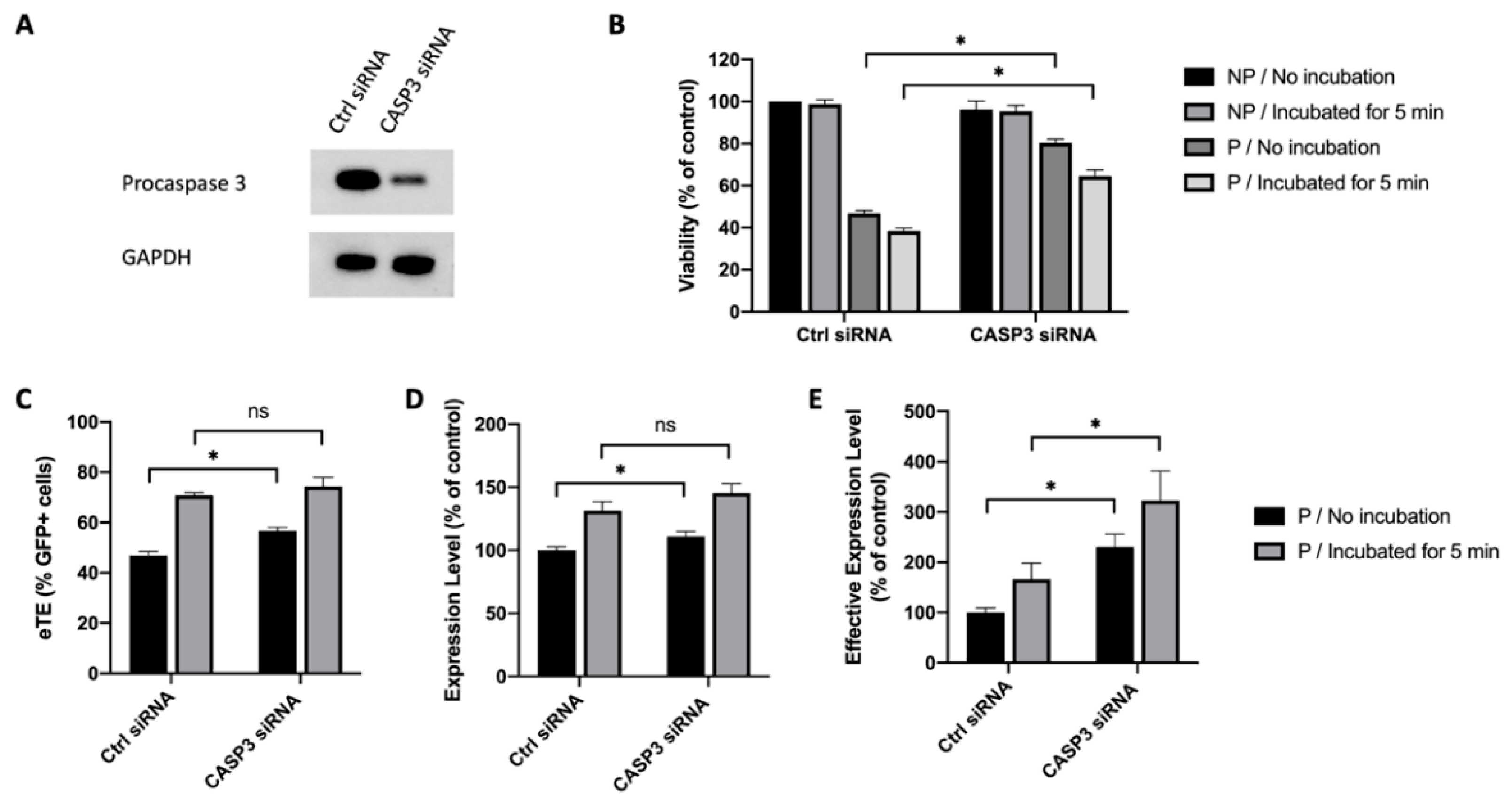
2.3. Caspase 3 Is a Key Mediator in Electrotransfer-Induced Cell Apoptosis
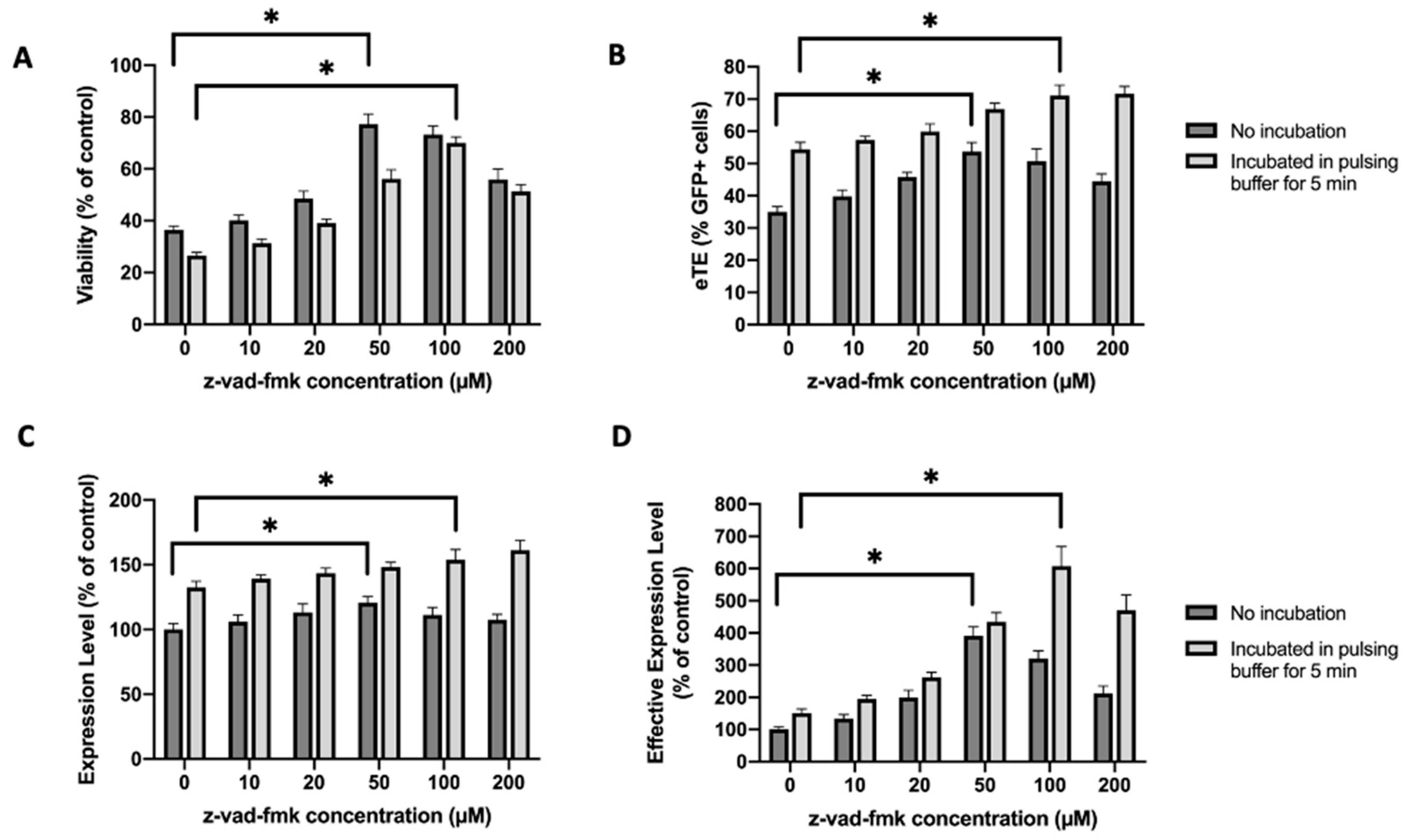
2.4. Similar Effects of Caspase Inhibition Are Observed in NIH/3T3 Fibroblasts
2.5. Inhibition of Apoptosis Improves Gene Editing Efficiency
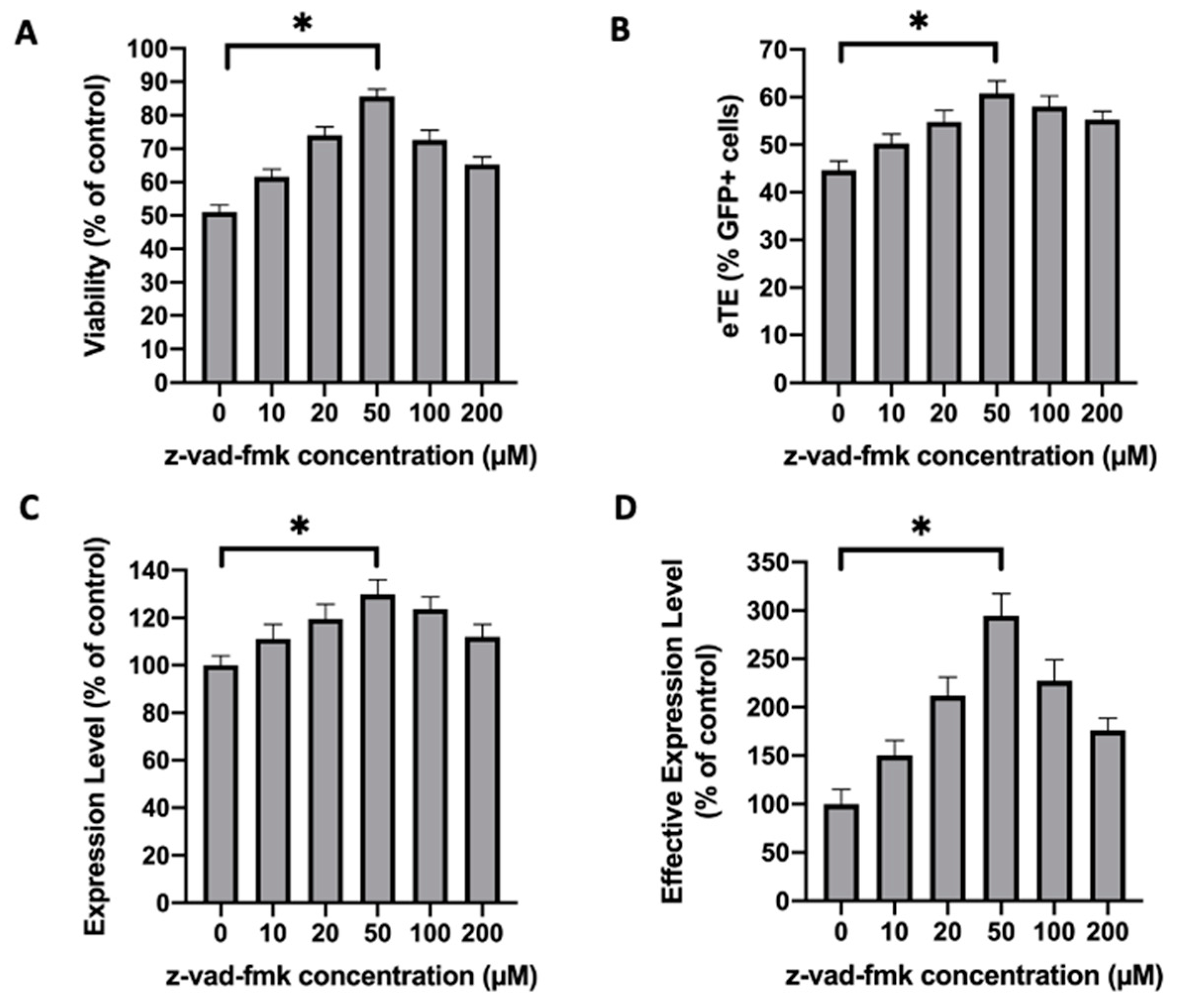
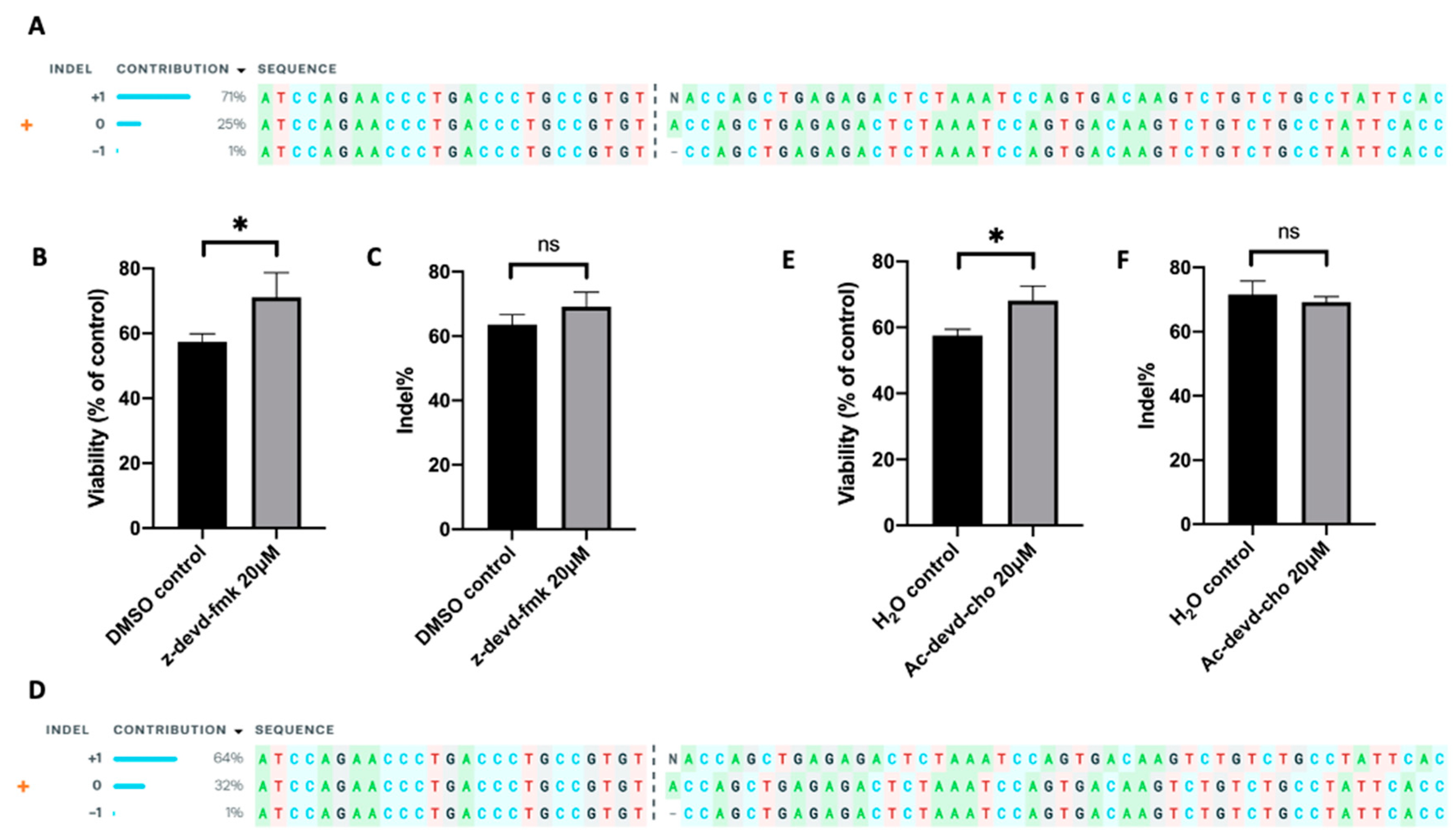
2.6. Inhibition of Caspase 3 in Human Primary T Cells Improves Gene Editing Efficiency
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Electrotransfer of pDNA
4.3. Treatment of Cells with Apoptosis Inhibitors
4.4. siRNA Knockdown of Procaspase-3 Expression
4.5. Western Blot Analysis
4.6. Flow Cytometry Analysis
4.7. TRAC Knockout by Cas9/sgRNA RNP in Jurkat Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Models Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood J. Am. Soc. Hematol. 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhou, G.; Zhang, L.; Zhao, Q. Building potent chimeric antigen receptor T cells with CRISPR genome editing. Front. Immunol. 2019, 10, 456. [Google Scholar] [CrossRef]
- Morgan, N.V.; Goddard, S.; Cardno, T.S.; McDonald, D.; Rahman, F.; Barge, D.; Ciupek, A.; Straatman-Iwanowska, A.; Pasha, S.; Guckian, M. Mutation in the TCRα subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRαβ+ T cells. J. Clin. Investig. 2011, 121, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lee, B.; Lee, A.Y.-F.; Modzelewski, A.J.; He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J. Biol. Chem. 2016, 291, 14457–14467. [Google Scholar] [CrossRef] [Green Version]
- Cervia, L.D.; Yuan, F. Current progress in electrotransfection as a nonviral method for gene delivery. Mol. Pharm. 2018, 15, 3617–3624. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.T.; Collins, M.; Terefe, J.; Ugozzoli, L.; Rubio, T. Optimizing electroporation conditions in primary and other difficult-to-transfect cells. J. Biomol. Tech. JBT 2008, 19, 328. [Google Scholar] [PubMed]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.P.; Huntoon, C.J.; Graham, D.; McKean, D.J. The analysis of costimulatory receptor signaling cascades in normal T lymphocytes using in vitro gene transfer and reporter gene analysis. Nat. Med. 2001, 7, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Aksoy, B.A.; Czech, E.; Hammerbacher, J. Viable and efficient electroporation-based genetic manipulation of unstimulated human T cells. BioRxiv 2019, 466243. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, D.-Q. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol. Cancer Ther. 2010, 9, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Lademann, U.; Cain, K.; Gyrd-Hansen, M.; Brown, D.; Peters, D.; Jäättelä, M. Diarylurea compounds inhibit caspase activation by preventing the formation of the active 700-kilodalton apoptosome complex. Mol. Cell. Biol. 2003, 23, 7829–7837. [Google Scholar] [CrossRef] [Green Version]
- Liskovykh, M.; Lee, N.C.; Larionov, V.; Kouprina, N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 2016, 3, 16043. [Google Scholar] [CrossRef]
- Maiwulanjiang, M.; Bi, C.W.; Lee, P.S.; Xin, G.; Miernisha, A.; Lau, K.M.; Xiong, A.; Li, N.; Dong, T.T.; Aisa, H.A. The volatile oil of Nardostachyos Radix et Rhizoma induces endothelial nitric oxide synthase activity in HUVEC cells. PLoS ONE 2015, 10, e0116761. [Google Scholar] [CrossRef]
- Dai, J.; Huang, Y.-J.; He, X.; Zhao, M.; Wang, X.; Liu, Z.-S.; Xue, W.; Cai, H.; Zhan, X.-Y.; Huang, S.-Y. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 2019, 176, 1447–1460.e1414. [Google Scholar] [CrossRef] [Green Version]
- Leclere, L.; Fransolet, M.; Cote, F.; Cambier, P.; Arnould, T.; Van Cutsem, P.; Michiels, C. Heat-modified citrus pectin induces apoptosis-like cell death and autophagy in HepG2 and A549 cancer cells. PLoS ONE 2015, 10, e0115831. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Wang, L.; Chang, C.-C.; Rothenberg, K.E.; Huang, J.; Wang, Y.; Hoffman, B.D.; Liton, P.B.; Yuan, F. Involvement of a Rac1-dependent macropinocytosis pathway in plasmid DNA delivery by electrotransfection. Mol. Ther. 2017, 25, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Beebe, S.J.; Sain, N.M.; Ren, W. Induction of cell death mechanisms and apoptosis by nanosecond pulsed electric fields (nsPEFs). Cells 2013, 2, 136–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuccitelli, R.; McDaniel, A.; Anand, S.; Cha, J.; Mallon, Z.; Berridge, J.C.; Uecker, D. Nano-Pulse Stimulation is a physical modality that can trigger immunogenic tumor cell death. J. Immunol. Ther. Cancer 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagalkot, T.R.; Terhune, R.C.; Leblanc, N.; Craviso, G.L. Different membrane pathways mediate Ca2+ influx in adrenal chromaffin cells exposed to 150–400 ns electric pulses. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.G.; Cullen, S.P.; Sheridan, C.; Lüthi, A.U.; Gerner, C.; Martin, S.J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA 2008, 105, 12815–12819. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a026716. [Google Scholar] [CrossRef] [Green Version]
- Yim, H.; Hwang, I.S.; Choi, J.-S.; Chun, K.-H.; Jin, Y.H.; Ham, Y.-M.; Lee, K.Y.; Lee, S.K. Cleavage of Cdc6 by caspase-3 promotes ATM/ATR kinase–mediated apoptosis of HeLa cells. J. Cell Biol. 2006, 174, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Shi, Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Bosnjak, M.; Jesenko, T.; Kamensek, U.; Sersa, G.; Lavrencak, J.; Heller, L.; Cemazar, M. Electrotransfer of different control plasmids elicits different antitumor effectiveness in B16. F10 melanoma. Cancers 2018, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-Y.; Libardo, M.D.J.; Angeles-Boza, A.M.; Pellois, J.-P. Membrane oxidation in cell delivery and cell killing applications. ACS Chem. Biol. 2017, 12, 1170–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golzio, M.; Gabriel, B.; Boissier, F.; Deuwille, J.; Rols, M.; Teissie, J. Calcium et cellules électropermeabilisées. J. De La Soc. De Biol. 2003, 197, 301–310. [Google Scholar] [CrossRef]
- Gabriel, B.; Teissie, J. Generation of reactive-oxygen species induced by electropermeabilization of Chinese hamster ovary cells and their consequence on cell viability. Eur. J. Biochem. 1994, 223, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Cemazar, M.; Heller, L.C. Cytosolic DNA sensor upregulation accompanies DNA electrotransfer in B16. F10 melanoma cells. Mol. Ther. Nucleic Acids 2016, 5, e322. [Google Scholar] [CrossRef] [Green Version]
- Chicaybam, L.; Sodre, A.L.; Curzio, B.A.; Bonamino, M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS ONE 2013, 8, e60298. [Google Scholar] [CrossRef]
- Muller, R.Y.; Hammond, M.C.; Rio, D.C.; Lee, Y.J. An Efficient method for electroporation of small interfering RNAs into ENCODE project tier 1 GM12878 and K562 cell lines. J. Biomol. Tech. JBT 2015, 26, 142. [Google Scholar] [CrossRef]
- Chang, C.-C.; Wu, M.; Yuan, F. Role of specific endocytic pathways in electrotransfection of cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14058. [Google Scholar] [CrossRef]
- Hsiau, T.; Conant, D.; Maures, T.; Waite, K.; Yang, J.; Kelso, R.; Holden, K.; Enzmann, B.L.; Stoner, R. Inference of CRISPR Edits from Sanger Trace Data. BioRxiv 2019. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Chang, C.-C.; Wang, L.; Yuan, F. Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing. Cancers 2020, 12, 2603. https://doi.org/10.3390/cancers12092603
Wang C, Chang C-C, Wang L, Yuan F. Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing. Cancers. 2020; 12(9):2603. https://doi.org/10.3390/cancers12092603
Chicago/Turabian StyleWang, Chunxi, Chun-Chi Chang, Liangli Wang, and Fan Yuan. 2020. "Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing" Cancers 12, no. 9: 2603. https://doi.org/10.3390/cancers12092603
APA StyleWang, C., Chang, C. -C., Wang, L., & Yuan, F. (2020). Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing. Cancers, 12(9), 2603. https://doi.org/10.3390/cancers12092603