
Drug Development in Tissue-Agnostic Indications



Abstract
:Simple Summary
Abstract
1. Introduction
2. Efficacy Endpoints: A Historical Perspective
3. Ancestral Paradigm of Drug Approval in Oncology
4. Impact of the Molecular Segmentation of Cancer
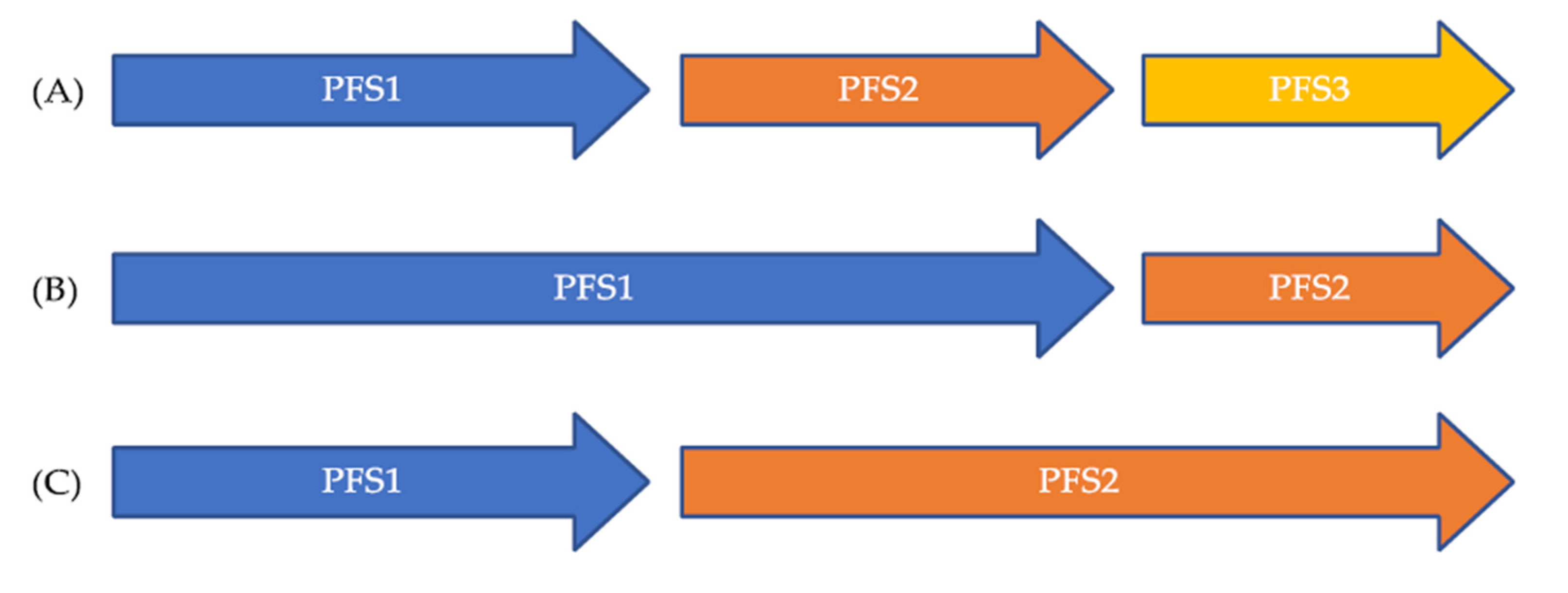
5. The PFS Ratio as a Novel Endpoint to Overcome Patient Heterogeneity
6. Review of Clinical Trials Using Each Patient as His/Her Own Control to Assess Drug Efficacy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zubrod, C.G.; Schneiderman, M.; Frei, E.; Brindley, C.; Lennard Gold, G.; Shnider, B.; Oviedo, R.; Gorman, J.; Jones, R.; Jonsson, U.; et al. Appraisal of Methods for the Study of Chemotherapy of Cancer in Man: Comparative Therapeutic Trial of Nitrogen Mustard and Triethylene Thiophosphoramide. J. Chronic Dis. 1960, 11, 7–33. [Google Scholar] [CrossRef]
- Wintrobe, M.M.; Huguley, C.M.; McLennan, M.T.; Penna de Carvalho Lima, L. Nitrogen Mustard as a Therapeutic Agent for Hodgkin’s Disease, Lymphosarcoma and Leukemia. Ann. Intern. Med. 1947, 27, 529. [Google Scholar] [CrossRef]
- Karnofsky, D.A.; Abelmann, W.H.; Craver, L.F.; Burchenal, J.H. The Use of the Nitrogen Mustards in the Palliative Treatment of Carcinoma. With Particular Reference to Bronchogenic Carcinoma. Cancer 1948, 634–656. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. PD-L1, TMB, MSI, and Other Predictors of Response to Immune Checkpoint Inhibitors in Biliary Tract Cancer. Cancers 2021, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Benayed, R.; Hechtman, J.F.; Ladanyi, M. Identifying Patients with NTRK Fusion Cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, M.; Eberhardt, W.E.E.; Hoffknecht, P.; Metzenmacher, M.; Wehler, T.; Kokowski, K.; Alt, J.; Schütte, W.; Büttner, R.; Heukamp, L.C.; et al. KRAS G12C-Mutated Advanced Non-Small Cell Lung Cancer: A Real-World Cohort from the German Prospective, Observational, Nation-Wide CRISP Registry (AIO-TRK-0315). Lung Cancer 2021, 154, 51–61. [Google Scholar] [CrossRef]
- Pillai, R.N.; Behera, M.; Berry, L.D.; Rossi, M.R.; Kris, M.G.; Johnson, B.E.; Bunn, P.A.; Ramalingam, S.S.; Khuri, F.R. HER2 Mutations in Lung Adenocarcinomas: A Report from the Lung Cancer Mutation Consortium: HER2 Mutations in Lung Adenocarcinomas. Cancer 2017, 123, 4099–4105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, J.G.; Otterson, G.A. Agents to Treat BRAF-Mutant Lung Cancer. Drugs Context 2019, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET -Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and Characterization of Microsatellite Instability across 18 Cancer Types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Krebs, M.G.; Le Tourneau, C.; Sokol, E.S.; Maund, T.R.; Wilson, T.R.; Jin, D.X.; Newberg, J.Y.; Fabrizio, D.; Veronese, L.; et al. Genomic Context of NTRK1/2/3 Fusion-Positive Tumours from a Large Real-World Population. Npj Precis. Oncol. in press.
- Breast Cancer Task Force Treatment Committee National Cancer Institute. Breast Cancer: Suggested Protocol Guidelines for Combination Chemotherapy Trials and for Combined Modality Trials. US Dep. Health Educ. Welf. Publ. NoNIH 1977, 77–1192. [Google Scholar]
- Hayward, J.; Carbone, P.P.; Heuson, J.-C.; Kumaoka, S.; Segaloff, A.; Rubens, R.D. Assessment of Response to Therapy in Advanced Breast Cancer. A Project of the Programme on Clinical Oncology of the International Union against Cancer, Geneva, Switzerland. Cancer 1977, 39, 6. [Google Scholar] [CrossRef]
- World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment; World Health Organization: Geneva, Switzerland, 1979. [Google Scholar]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Glabbeke, M.V.; van Oosterom, T.; Christian, M.C.; et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel Patterns of Response under Immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gangadhar, T.C.; et al. Evaluation of Immune-Related Response Criteria and RECIST v1.1 in Patients With Advanced Melanoma Treated With Pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. IRECIST: Guidelines for Response Criteria for Use in Trials Testing Immunotherapeutics. Lancet Oncol. 2017, 18, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Frelaut, M.; du Rusquec, P.; de Moura, A.; Le Tourneau, C.; Borcoman, E. Pseudoprogression and Hyperprogression as New Forms of Response to Immunotherapy. BioDrugs 2020, 34, 463–476. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Facts: Biomarkers and Surrogate Endpoints; FDA: Rockwell,, ML, USA, 2019. [Google Scholar]
- Chen, E.Y.; Joshi, S.K.; Tran, A.; Prasad, V. Estimation of Study Time Reduction Using Surrogate End Points Rather Than Overall Survival in Oncology Clinical Trials. JAMA Intern. Med. 2019, 179, 642. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, F.; Leonardo, G.D.; Pino, M.S.; Martella, F.; Perboni, S.; Ribecco, A.; Fioretto, L. Progression-Free Survival as a Surrogate End-Point in Advanced Colorectal Cancer Treated with Antiangiogenic Therapies. Anticancer Res. 2016, 36, 4259–4265. [Google Scholar] [PubMed]
- Kim, C.; Prasad, V. Cancer Drugs Approved on the Basis of a Surrogate End Point and Subsequent Overall Survival: An Analysis of 5 Years of US Food and Drug Administration Approvals. JAMA Intern. Med. 2015, 175, 1992–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushti, S.L.; Mulkey, F.; Sridhara, R. Evaluation of Overall Response Rate and Progression-Free Survival as Potential Surrogate Endpoints for Overall Survival in Immunotherapy Trials. Clin. Cancer Res. 2018, 24, 2268–2275. [Google Scholar] [CrossRef] [Green Version]
- Van Wilpe, S.; Koornstra, R.; Den Brok, M.; De Groot, J.W.; Blank, C.; De Vries, J.; Gerritsen, W.; Mehra, N. Lactate Dehydrogenase: A Marker of Diminished Antitumor Immunity. OncoImmunology 2020, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T.K. S100B and LDH as Early Prognostic Markers for Response and Overall Survival in Melanoma Patients Treated with Anti-PD-1 or Combined Anti-PD-1 plus Anti-CTLA-4 Antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum Lactate Dehydrogenase as an Early Marker for Outcome in Patients Treated with Anti-PD-1 Therapy in Metastatic Melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Yin, C.; Jiang, C.; Liao, F.; Rong, Y.; Cai, X.; Guo, G.; Qiu, H.; Chen, X.; Zhang, B.; He, W.; et al. Initial LDH Level Can Predict the Survival Benefit from Bevacizumab in the First-Line Setting in Chinese Patients with Metastatic Colorectal Cancer. OncoTargets Ther. 2014, 1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.M.; Rowland, A.; Kichenadasse, G.; Wiese, M.D.; Gurney, H.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Predicting Response and Toxicity to Immune Checkpoint Inhibitors Using Routinely Available Blood and Clinical Markers. Br. J. Cancer 2017, 117, 913–920. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA Approval Summary: Crizotinib for the Treatment of Metastatic Non-Small Cell Lung Cancer With Anaplastic Lymphoma Kinase Rearrangements. Oncologist 2014, 19, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Kwak, E.L.; Shaw, A.T.; Ou, S.-H.I.; Varella-Garcia, M.; Stubbs, H.; Gandhi, L.; Ratain, M.J.; Wilner, K.; Iafrate, A.J. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in Advanced ALK -Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, L.A.; Le, D.T.; Yoshino, T.; Andre, T.; Bendell, J.C.; Koshiji, M.; Zhang, Y.; Kang, S.P.; Lam, B.; Jäger, D. KEYNOTE-177: Randomized Phase III Study of Pembrolizumab versus Investigator-Choice Chemotherapy for Mismatch Repair-Deficient or Microsatellite Instability-High Metastatic Colorectal Carcinoma. J. Clin. Oncol. 2017, 35, TPS815. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves Landmark Tissue-Agnostic Cancer Drug. Nat. Rev. Drug Discov. 2019, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Donoghue, M.; Aungst, S.; Myers, C.E.; Helms, W.S.; Shen, G.; Zhao, H.; Stephens, O.; Keegan, P.; Pazdur, R. FDA Approval Summary: Entrectinib for the Treatment of NTRK Gene Fusion Solid Tumors. Clin. Cancer Res. 2021, 27, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.J.; Zehir, A.; Sireci, A.N.; Aisner, D.L. Detection of Tumor NTRK Gene Fusions to Identify Patients Who May Benefit from Tyrosine Kinase (TRK) Inhibitor Therapy. J. Mol. Diagn. 2019, 21, 553–571. [Google Scholar] [CrossRef] [Green Version]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and Drug-Sensitive NTRK1 Rearrangements in Lung Cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Patel, M.R.; Bauer, T.M.; Liu, S.V.; Drilon, A.E.; Wheler, J.J.; Shaw, A.T.; Farago, A.F.; Ou, S.-H.I.; Luo, D.; Yeh, L.; et al. STARTRK-1: Phase 1/2a Study of Entrectinib, an Oral Pan-Trk, ROS1, and ALK Inhibitor, in Patients with Advanced Solid Tumors with Relevant Molecular Alterations. J. Clin. Oncol. 2015, 33, 2596. [Google Scholar] [CrossRef]
- Drilon, A.; Sankhala, K.K.; Liu, S.V.; Cho, B.C.; Blakely, C.; Chee, C.E.; Fakih, M.; Polikoff, J.; Hornby, Z.; Schechet, L.; et al. Abstract CT060: STARTRK-2: A Global Phase 2, Open-Label, Basket Study of Entrectinib in Patients with Locally Advanced or Metastatic Solid Tumors Harboring TRK, ROS1, or ALK Gene Fusions. Cancer Res. 2017, 77, CT060. [Google Scholar] [CrossRef]
- De Braud, F.G.; Niger, M.; Damian, S.; Bardazza, B.; Martinetti, A.; Pelosi, G.; Marrapese, G.; Palmeri, L.; Cerea, G.; Valtorta, E.; et al. Alka-372-001: First-in-Human, Phase I Study of Entrectinib—An Oral Pan-Trk, ROS1, and ALK Inhibi—In Patients with Advanced Solid Tumors with Relevant Molecular Alterations. J. Clin. Oncol. 2015, 33, 2517. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Stephenson, J.J.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Kamal, M.; Tsimberidou, A.-M.; Bedard, P.; Pierron, G.; Callens, C.; Rouleau, E.; Vincent-Salomon, A.; Servant, N.; Alt, M.; et al. Treatment Algorithms Based on Tumor Molecular Profiling: The Essence of Precision Medicine Trials. J. Natl. Cancer Inst. 2016, 108, djv362. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly Targeted Therapy Based on Tumour Molecular Profiling versus Conventional Therapy for Advanced Cancer (SHIVA): A Multicentre, Open-Label, Proof-of-Concept, Randomised, Controlled Phase 2 Trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Chen, A.P.; Kummar, S.; Moore, N.; Rubinstein, L.V.; Zhao, Y.; Williams, P.M.; Palmisano, A.; Sims, D.; Coyne, G.O.; Rosenberger, C.L.; et al. Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT): A Randomized Multicenter Phase II Trial. JCO Precis. Oncol. 2021, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular Profiling of Cancer Patients Enables Personalized Combination Therapy: The I-PREDICT Study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and Transcriptomic Profiling Expands Precision Cancer Medicine: The WINTHER Trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Belin, L.; Kamal, M.; Mauborgne, C.; Plancher, C.; Mulot, F.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; et al. Randomized Phase II Trial Comparing Molecularly Targeted Therapy Based on Tumor Molecular Profiling versus Conventional Therapy in Patients with Refractory Cancer: Cross-over Analysis from the SHIVA Trial. Ann. Oncol. 2017, 28, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF -Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in Non-Small-Cell Lung Cancer Patients with BRAFV600 and BRAFnonV600 Mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF -Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.-P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF -Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Endpoint | Start | Event | Censored Events | Pros | Cons |
|---|---|---|---|---|---|
| PFS (Progression-Free Survival) | Randomization | Disease progression; Death | Last date of radiological assessment | Earlier read out Effect on survival not diluted by subsequent therapies | Death might not be related to disease but to comorbidities |
| TTP (Time to Progression) | Randomization | Disease progression | Death | Earlier read out Effect on survival not diluted by subsequent therapies | Does not take into account death related to treatments |
| TTF (Time to Treatment Failure) | Randomization | Disease progression; Adverse events; Patient choice; Death | - | Earlier read out Effect on survival not diluted by subsequent therapies Best indicator of the treatment’s specific tolerance | Does not completely reflect the duration of treatment efficacy, since patients might still be responding to treatment although they had an adverse event or decided to stop treatment Death might not be related to disease but to comorbidities |
| FFS (Failure-Free Survival) | The first day of treatment | Disease progression | Adverse event Death | Earlier read out Effect on survival not diluted by subsequent therapies Best indicator of the treatment’s specific efficacy | Death and adverse events related to treatments are not taken into account |
| DOR (Duration of Response) | Date of response | Disease progression | Death | Measures duration of response that might be of importance for some treatments such as immunohterapies | Only applies to the responding patient population |
| OS (Overall Survival) | Randomization | Death | Last date patient seen alive | Direct measure of clinical benefit, Easily measured, Gold standard endpoint | affected by post-progression and cross over therapies; require prolonged Follow up; includes non-cancer related deaths |
| Study | Von Hoff’s Study [49] | SHIVA01 [51,56] | MOSCATO-01 [53] | I-PREDICT [54] | WINTHER [55] |
|---|---|---|---|---|---|
| Threshold used for PFS ratio | 1.3 | 1.3 | 1.3 | 1.3 | 1.5 |
| No. of patients included | 106 | 741 | 1035 | 149 | 303 |
| No. of patients treated with matched therapy (%) | 66 (62%) | 170 (23%) | 199 (19%) | 73 (49%) | 107 (35%) |
| No. of evaluable patients for the PFS ratio (%) | 66 (62%) | 95 (13%) | 193 (19%) | 53 (36%) | 107 (35%) |
| Proportion of patient with a PFS ratio >1.3 | 27% | 37% 1 61% 2 | 33% | 45% | 25% |
| Median PFS1 (months) | - | 2.0 1 2.3 2 | - | - | - |
| Median PFS2 (months) | - | 2.1 1 2.8 2 | 2.3 | 3.7 | 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
du Rusquec, P.; Le Tourneau, C. Drug Development in Tissue-Agnostic Indications. Cancers 2021, 13, 2758. https://doi.org/10.3390/cancers13112758
du Rusquec P, Le Tourneau C. Drug Development in Tissue-Agnostic Indications. Cancers. 2021; 13(11):2758. https://doi.org/10.3390/cancers13112758
Chicago/Turabian Styledu Rusquec, Pauline, and Christophe Le Tourneau. 2021. "Drug Development in Tissue-Agnostic Indications" Cancers 13, no. 11: 2758. https://doi.org/10.3390/cancers13112758
APA Styledu Rusquec, P., & Le Tourneau, C. (2021). Drug Development in Tissue-Agnostic Indications. Cancers, 13(11), 2758. https://doi.org/10.3390/cancers13112758