
VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment


 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Transfection
2.3. Mitochondrial Membrane Potential (ΔΨ) and Cellular ATP Levels
2.4. Xenograft Experiments
2.5. Sirius Red Staining
2.6. Immunohistochemistry (IHC) and Immunofluorescence (IF) Staining of Tumor Tissue Sections
2.7. Tumor Tissue Extracts and Immunoblotting
2.8. RNA Preparation and Quantitative RT-PCR (q-RT-PCR)
2.9. NGS and Bioinformatics Analyses
2.10. Statistics
3. Results
3.1. si-hVDAC1-2A Inhibits Tumor Growth and Reduces Energy Production in a Lung Cancer Xenograft Model
3.2. Next-Generation Sequencing (NGS) of si-NT- and si-hVDAC1-2A-TTs Reveals Changes in the Expression of Mouse ECM Structure-Related Genes upon si-hVDAC1-2A Treatment
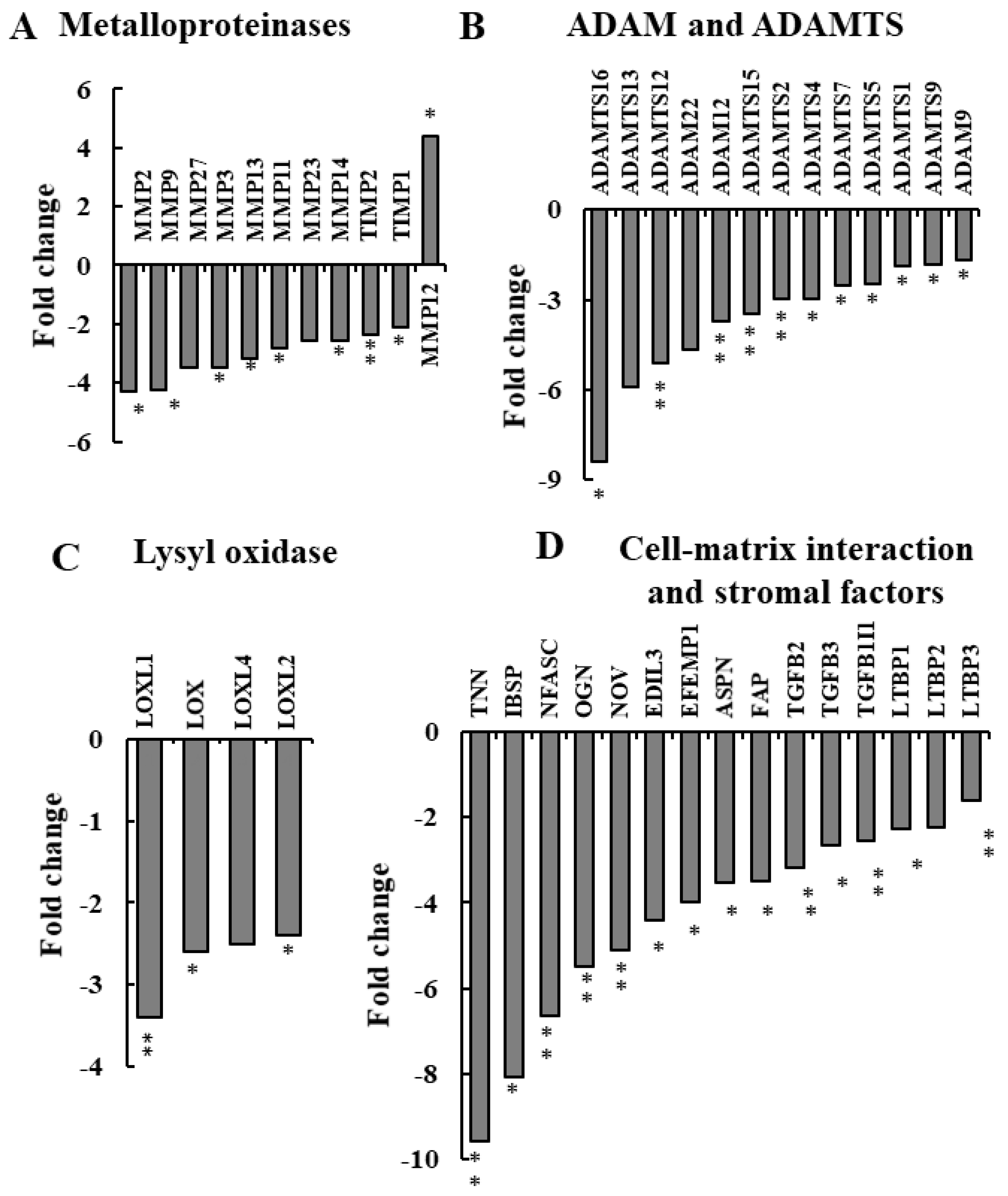
3.3. si-hVDAC1-2A Alters ECM Organization-Related Genes
3.4. NGS Analysis of si-NT-TTs and si-hVDAC1-2A-TTs Reveals Reduced Expression of ECM Deposition- and Degradation-Related Genes
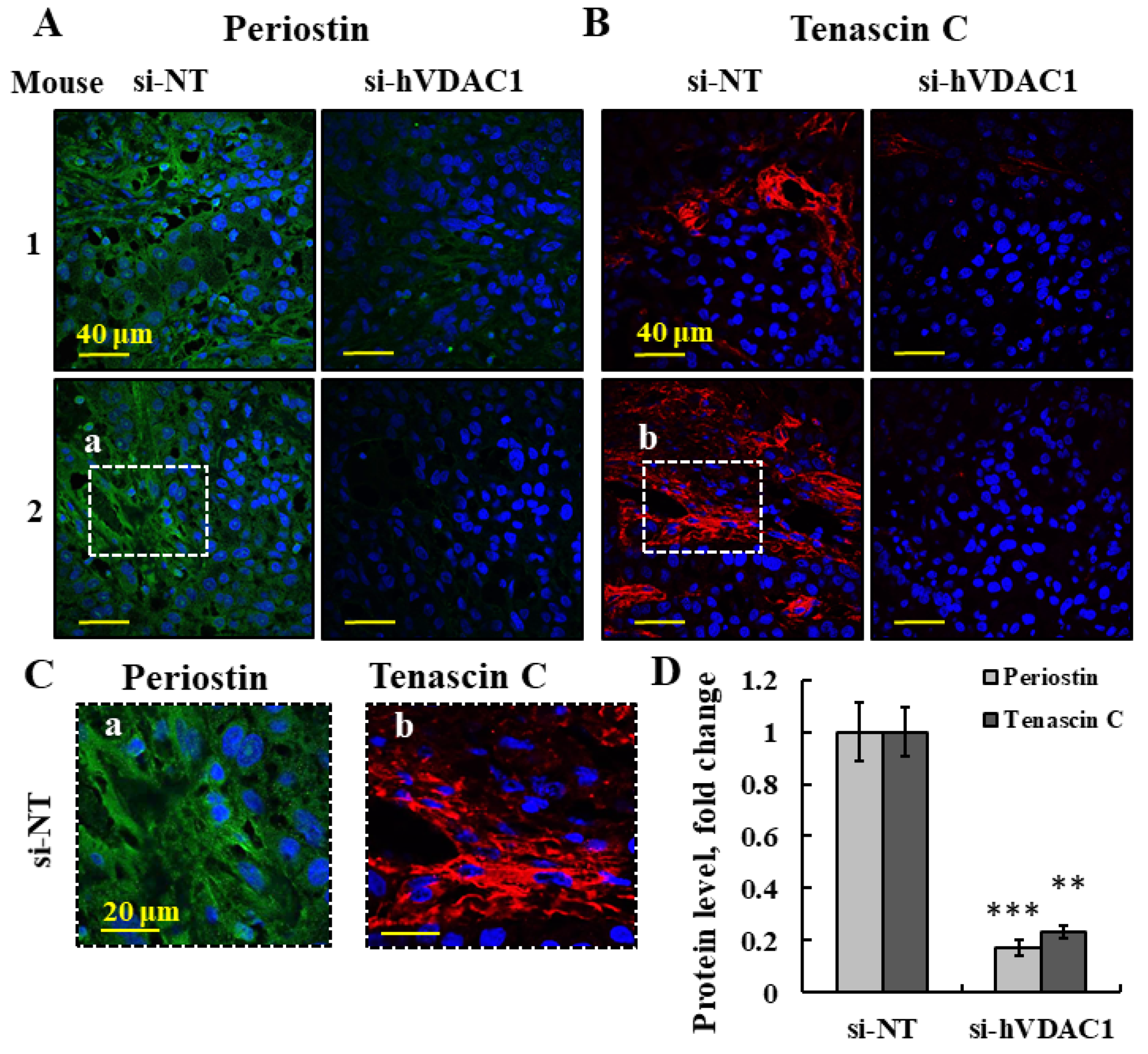
3.5. hVDAC1 Depletion in a Tumor Changes ECM Organization and Collagen Levels by Altering the Ability of Activated Fibroblasts to Encapsulate Cancer Cells in the Tumor
3.6. Tumor Treatment with si-hVDAC1-2A Altered the Expression of Mouse Genes Associated with Angiogenesis of TME Cells
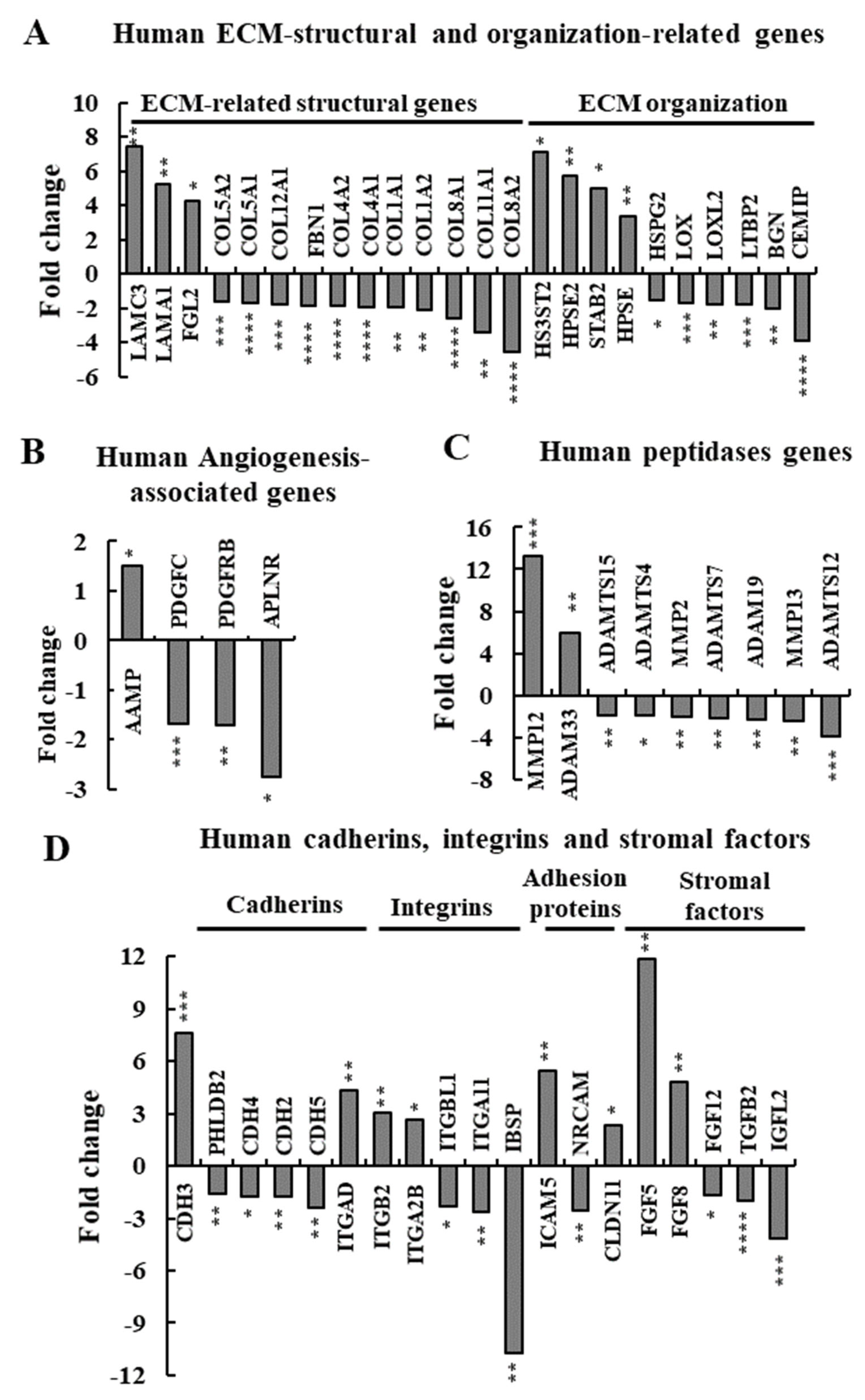
3.7. si-hVDAC1-2A Effects on the Expression of TME-Related Human Genes
3.8. Validation of the Effects of si-hVDAC1-2A on Key Mouse TME Factors by q-RT-PCR
4. Discussion
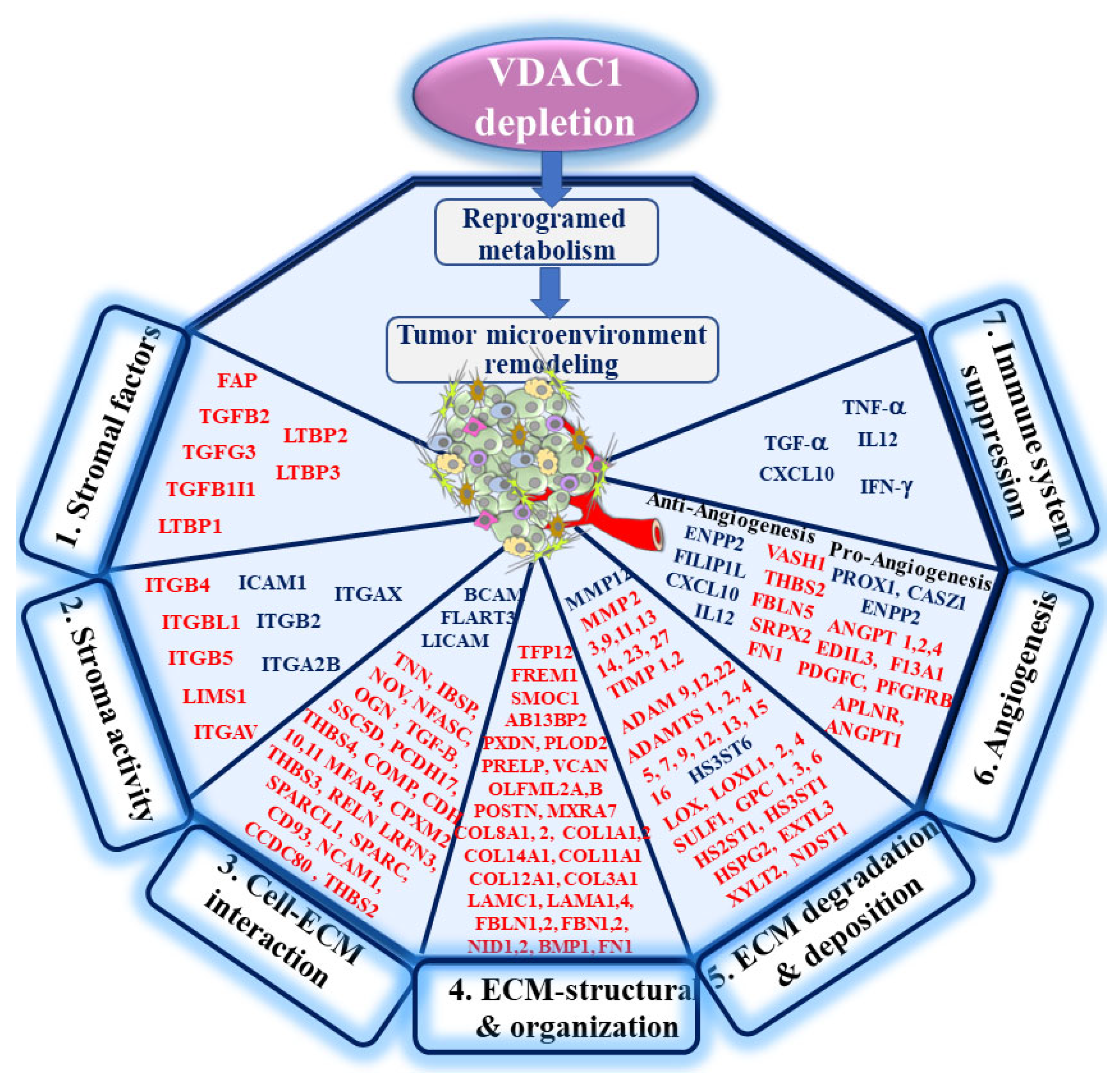
4.1. The Link between Reprogramed Cancer Cell Metabolism and the TME
4.2. hVDAC1 Depletion Alters the Expression of ECM-Related Proteins and Stromal Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Asp. Med. 2010, 31, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, I. Therapeutic potential of targeting glucose metabolism in glioma stem cells. Expert Opin. Ther. Targets 2014, 18, 1233–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. The Tumor-Promoting Flow of Cells Into, Within and Out of the Tumor Site: Regulation by the Inflammatory Axis of TNFalpha and Chemokines. Cancer Microenviron. 2012, 5, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Bar, J.O.A.; Herbst, R.S. Molecular Events Surrounding the Angiogenic Switch of Lung Cancer. In Principles and Practice of Lung Cancer, 4th ed.; Pass, H.I., Carbone, D.P., Johnson, D.H., Minna, J.D., Scagliotti, G.V., Turrisi, A.T., III, Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2010; pp. 113–134. [Google Scholar]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J. Good neighbours in the tumour stroma reduce oxidative stress. Nat. Cell Biol. 2012, 14, 235–236. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Bazellieres, E.; Allen, M.D.; Andreu, I.; Oria, R.; Sunyer, R.; Gomm, J.J.; Marshall, J.F.; Jones, J.L.; Trepat, X.; et al. Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 2014, 13, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force Fluctuations within Focal Adhesions Mediate ECM-Rigidity Sensing to Guide Directed Cell Migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Witz, I.P.; Levy-Nissenbaum, O. The tumor microenvironment in the post-PAGET era. Cancer Lett. 2006, 242, 1–10. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Sporn, M.B. The war on cancer. Lancet 1996, 347, 1377–1381. [Google Scholar] [CrossRef]
- Yan, W.; Shao, R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J. Biol. Chem. 2006, 281, 19700–19708. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Siriwardena, B.S.; Hatano, H.; Ogawa, I.; Takata, T. Periostin: Novel diagnostic and therapeutic target for cancer. Histol. Histopathol. 2007, 22, 1167–1174. [Google Scholar] [CrossRef]
- Kyutoku, M.; Taniyama, Y.; Katsuragi, N.; Shimizu, H.; Kunugiza, Y.; Iekushi, K.; Koibuchi, N.; Sanada, F.; Oshita, Y.; Morishita, R. Role of periostin in cancer progression and metastasis: Inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int. J. Mol. Med. 2011, 28, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, F.S.; Jones, P.L. The tenascin family of ECM glycoproteins: Structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. 2000, 218, 235–259. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Chiquet, M. Tenascins: Regulation and putative functions during pathological stress. J. Pathol. 2003, 200, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Porporato, P.E.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol. Life Sci. 2016, 73, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W. Mechanisms underlying hypoxia development in tumors. Adv. Exp. Med. Biol. 2003, 510, 51–56. [Google Scholar]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Yabu, M.; Shime, H.; Hara, H.; Saito, T.; Matsumoto, M.; Seya, T.; Akazawa, T.; Inoue, N. IL-23-dependent and -independent enhancement pathways of IL-17A production by lactic acid. Int. Immunol. 2011, 23, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfor, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.; Sheiko, T.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. BBA Biomembranes 2012, 1818, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 2020, 12, 1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther. Nucleic Acids 2019, 17, 24–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro. Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. [Google Scholar] [CrossRef] [Green Version]
- Koren, I.; Raviv, Z.; Shoshan-Barmatz, V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. Ther. 2010, 9, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Shteinfer-Kuzmine, A.; Verma, A.; Arif, T.; Aizenberg, O.; Paul, A.; Shoshan-Barmaz, V. Mitochondria and nucleus cross-talk: Signaling in metabolism, apoptosis, and differentiation, and function in cancer. IUBMB Life 2021, 73, 492–510. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1889, 8, 98–101. [Google Scholar]
- Junqueira, L.C.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Krelin, Y.; Arif, T.; Jeger, R.; Shoshan-Barmatz, V. A New Role for the Mitochondrial Pro-apoptotic Protein SMAC/Diablo in Phospholipid Synthesis Associated with Tumorigenesis. Mol. Ther. 2018, 26, 680–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Gatenby, R.A. Adaptive landscapes and emergent phenotypes: Why do cancers have high glycolysis? J. Bioenerg. Biomembr. 2007, 39, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Majeed, R.H.A.; Qurishi, Y.; Qazi, A.K.; Hussain, A.; Ahmed, M.; Najar, R.A.; Bhat, J.A.; Singh, S.K.; Saxena, A.K. Therapeutic Targeting of Cancer Cell Metabolism: Role of Metabolic Enzymes, Oncogenes and Tumor Suppressor Genes. J. Cancer Sci. Ther. 2012, 4, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Daley, W.P.; Peters, S.B.; Larsen, M. Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 2008, 121, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Balcioglu, H.E.; Danen, E.H. Integrin signaling in control of tumor growth and progression. Int. J. Biochem. Cell Biol. 2013, 45, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.W. Functional role of periostin in development and wound repair: Implications for connective tissue disease. J. Cell Commun. Signal 2008, 2, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(v)/beta(3) and alpha(v)beta(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar] [PubMed]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal 2009, 3, 287–310. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-C and integrins in cancer. Cell Adhes. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, A.M.; Grisolano, J.L.; Baumann, M.L.; Kobayashi, D.K.; Hautamaki, R.D.; Nehring, L.C.; Cornelius, L.A.; Shapiro, S.D. Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res. 2006, 66, 6149–6155. [Google Scholar] [CrossRef] [Green Version]
- Acuff, H.B.; Sinnamon, M.; Fingleton, B.; Boone, B.; Levy, S.E.; Chen, X.W.; Pozzi, A.; Carbone, D.P.; Schwartz, D.R.; Moin, K.; et al. Analysis of host- and tumor-derived proteinases using a custom dual species microarray reveals a protective role for stromal matrix metal loproteinase-12 in non-small cell lung cancer. Cancer Res. 2006, 66, 7968–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelius, L.A.; Nehring, L.C.; Harding, E.; Bolanowski, M.; Welgus, H.G.; Kobayashi, D.K.; Pierce, R.A.; Shapiro, S.D. Matrix metalloproteinases generate angiostatin: Effects on neovascularization. J. Immunol. 1998, 161, 6845–6852. [Google Scholar]
- Liu, J.; Ping, W.; Zu, Y.; Sun, W. Correlations of lysyl oxidase with MMP2/MMP9 expression and its prognostic value in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6040–6047. [Google Scholar]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Gao, Y.J.; Xiao, Q.A.; Ma, H.M.; Li, L.; Liu, J.; Feng, Y.; Fang, Z.Y.; Wu, J.; Han, X.K.; Zhang, J.H.; et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 18892–18897. [Google Scholar] [CrossRef] [Green Version]
- Martina, E.; Chiquet-Ehrismann, R.; Brellier, F. Tenascin-W: An extracellular matrix protein associated with osteogenesis and cancer. Int. J. Biochem. Cell Biol. 2010, 42, 1412–1415. [Google Scholar] [CrossRef]
- Ogata, Y. Bone sialoprotein and its transcriptional regulatory mechanism. J. Periodontal Res. 2008, 43, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Board, R.; Jayson, G.C. Platelet-derived growth factor receptor (PDGFR): A target for anticancer therapeutics. Drug Resist Updat. 2005, 8, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.W.; Clair, T.; Kim, Y.S.; McMarlin, A.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001, 61, 6938–6944. [Google Scholar]
- Strieter, R.M.; Belperio, J.A.; Burdick, M.D.; Sharma, S.; Dubinett, S.M.; Keane, M.P. CXC chemokines—Angiogenesis, immunoangiostasis, and metastases in lung cancer. Ann. N. Y. Acad. Sci. 2004, 1028, 351–360. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Kunimasa, K.; Goto, T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. Int. J. Mol. Sci. 2020, 21, 597. [Google Scholar] [CrossRef] [Green Version]
- Midan, A.; Budhani, P.; Sheng, J.; Balasubramanyam, S.; Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Haria, D.D.; Curran, M.A. Tumor hypoxia drives immune suppression and immunotherapy resistance. J. Immunother. Cancer. 2015, 3, P392. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 1: Current Strategies and Agents. Pharm. Ther. 2017, 42, 375–383. [Google Scholar]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Baldini, N.; Sonveaux, P.; De Milito, A.; Supuran, C.T.; Otto, A.M.; Stock, C.M.; Pedersen, S.F.; Favicchio, R.; Avnet, S. Metabolism and microenvironment in cancer plasticity. Cancer Metab. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Morrot, A.; da Fonseca, L.M.; Salustiano, E.J.; Gentile, L.B.; Conde, L.; Filardy, A.A.; Franklim, T.N.; da Costa, K.M.; Freire-de-Lima, C.G.; Freire-de-Lima, L. Metabolic Symbiosis and immunomodulation: How Tumor Cell-Derived Lactate May Disturb innate and Adaptive immune Responses. Front. Oncol. 2018, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.T.; McLeod, K.; Kim, S.; Conway, S.J.; Hamilton, D.W. Periostin as a multifunctional modulator of the wound healing response. Cell Tissue Res. 2016, 365, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Sugihara, E.; Ohta, S.; Izuhara, K.; Funakoshi, T.; Amagai, M.; Saya, H. Periostin Is a Key Niche Component for Wound Metastasis of Melanoma. PLoS ONE 2015, 10, e0129704. [Google Scholar] [CrossRef]
- Brack, S.S.; Silacci, M.; Birchler, M.; Neri, D. Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C. Clin. Cancer Res. 2006, 12, 3200–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Syed, V. TGF-beta Signaling in Cancer. J. Cell Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.R.; Bae, Y.; Iwata, C.; Morishita, Y.; Yashiro, M.; Oka, M.; Fujii, T.; Komuro, A.; Kiyono, K.; Kaminishi, M.; et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 3460–3465. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Jechlinger, M.; Grunert, S.; Tamir, I.H.; Janda, E.; Ludemann, S.; Waerner, T.; Seither, P.; Weith, A.; Beug, H.; Kraut, N. Expression profiling of epithelial plasticity in tumor progression. Oncogene 2003, 22, 7155–7169. [Google Scholar] [CrossRef] [Green Version]
- Soltermann, A.; Tischler, V.; Arbogast, S.; Braun, J.; Probst-Hensch, N.; Weder, W.; Moch, H.; Kristiansen, G. Prognostic Significance of Epithelial-Mesenchymal and Mesenchymal-Epithelial Transition Protein Expression in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 7430–7437. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.M.; Lu, J.J.; Zhao, J.; Wei, X.; Wang, L.; Liu, P.S. Periostin Facilitates the Epithelial-Mesenchymal Transition of Endometrial Epithelial Cells through ILK-Akt Signaling Pathway. Biomed. Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanamura, N.; Yoshida, T.; Matsumoto, E.; Kawarada, Y.; Sakakura, T. Expression of fibronectin and tenascin-C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int. J. Cancer 1997, 73, 10–15. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, K.A.; Lopez, K.M. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. 2018, 417, 174–181. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Harris, B.F.; Berry, H.E.; Bergstrom, M.L.; Bronson, D.E.; Donahue, R.T. Nude 2 Jackson: A New Spontaneous Mutation in Foxn1; MGI Direct Data Submission: The Yews, UK, 2013. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Catalog Number | Company |
|---|---|---|
| In vivo JetPEI-Transfection reagent | 201-10G | PolyPlus (Illkirch, France) |
| JetPRIME- Transfection reagent | 114-15 | PolyPlus (Illkirch, France) |
| Triton X-100 | T-6878 | Sigma (St. Louis, MO, USA) |
| Tween-20 | 20452301 | Bio-Lab ltd. (Jerusalem, Israel) |
| Paraformaldehyde (PFA) | 15710 | Emsdiasum (Hatfield, PA, USA) |
| Dulbecco’s modified Eagle’s medium (DMEM) | 41965-039 | Gibco (Grand Island, NY, USA) |
| Roswell Park Memorial Institute (RPMI) 1640 | 21875-034 | Gibco (Grand Island, NY, USA) |
| Normal goat serum (NGS) | 04-009-1A | Biological Industries (Beit Haemek, Israel) |
| Fetal bovine serum (FBS) | 04-007-1A | Biological Industries (Beit Haemek, Israel) |
| L-glutamine | 03-020-1C | Biological Industries (Beit Haemek, Israel) |
| Penicillin–streptomycin | 03-031-5B | Biological Industries (Beit Haemek, Israel) |
| 3,3-diaminobenzidine (DAB) | SK-4105 | ImmPact-DAB (Burlingame, CA, USA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerbib, E.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment. Cancers 2021, 13, 2850. https://doi.org/10.3390/cancers13112850
Zerbib E, Arif T, Shteinfer-Kuzmine A, Chalifa-Caspi V, Shoshan-Barmatz V. VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment. Cancers. 2021; 13(11):2850. https://doi.org/10.3390/cancers13112850
Chicago/Turabian StyleZerbib, Erez, Tasleem Arif, Anna Shteinfer-Kuzmine, Vered Chalifa-Caspi, and Varda Shoshan-Barmatz. 2021. "VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment" Cancers 13, no. 11: 2850. https://doi.org/10.3390/cancers13112850
APA StyleZerbib, E., Arif, T., Shteinfer-Kuzmine, A., Chalifa-Caspi, V., & Shoshan-Barmatz, V. (2021). VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment. Cancers, 13(11), 2850. https://doi.org/10.3390/cancers13112850