
Coordinating Effect of VEGFC and Oleic Acid Participates to Tumor Lymphangiogenesis

 , , , ,
, , , ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
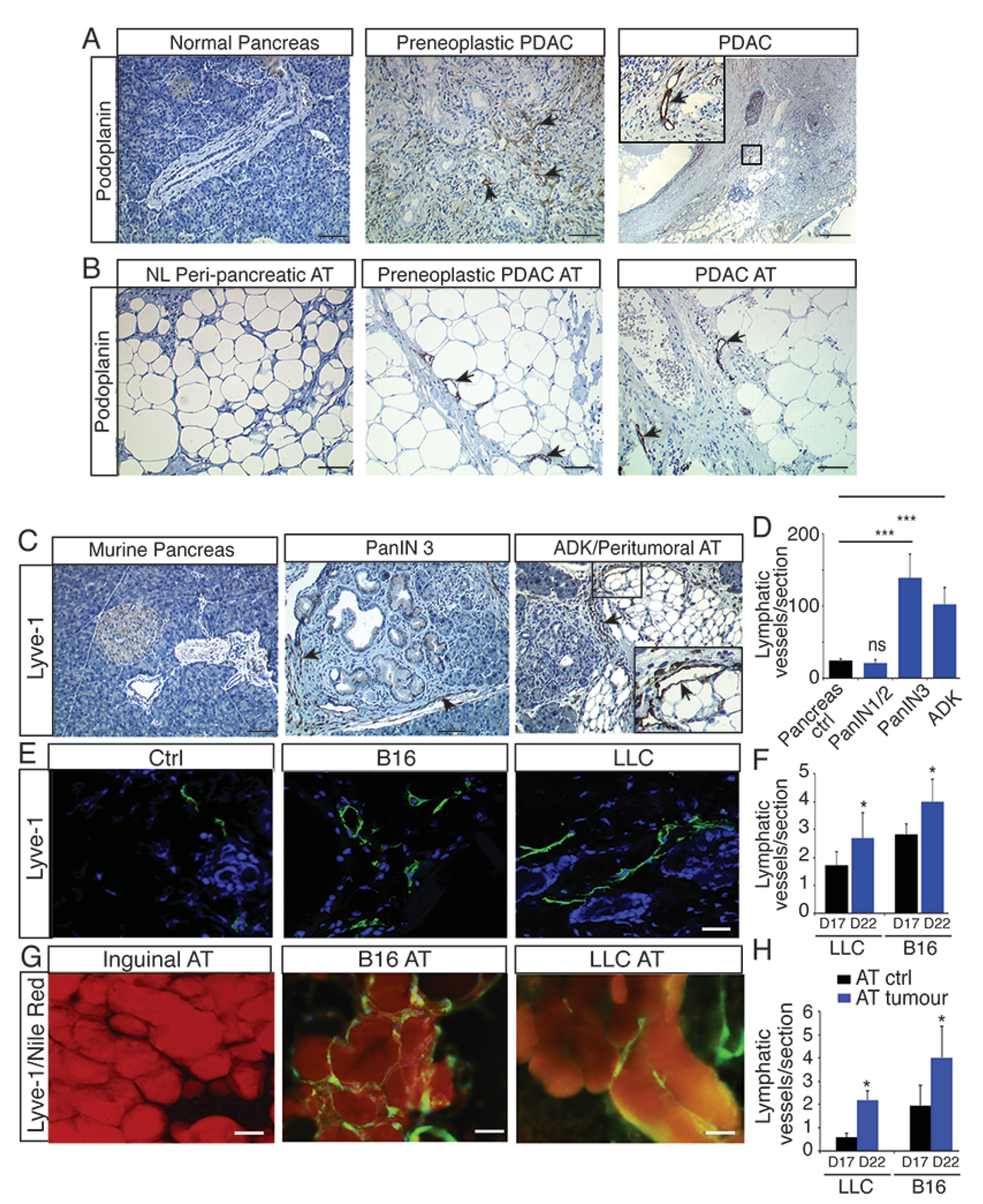
2.1. Lymphangiogenesis Develops in Preneoplastic Lesions and in Peritumoral Adipose Tissue
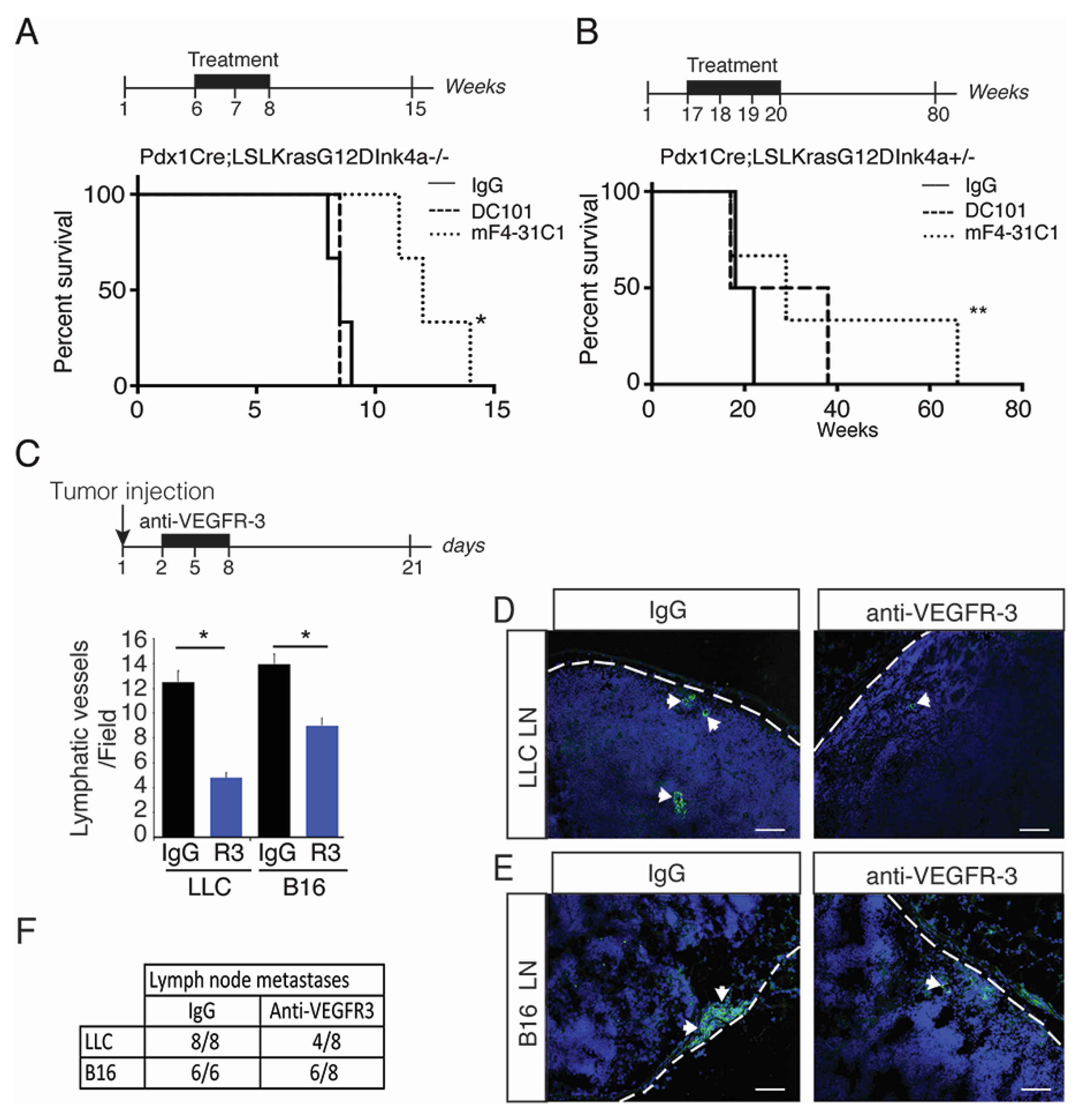
2.2. Blocking Preneoplastic Lesion Lymphangiogenesis Improves Survival
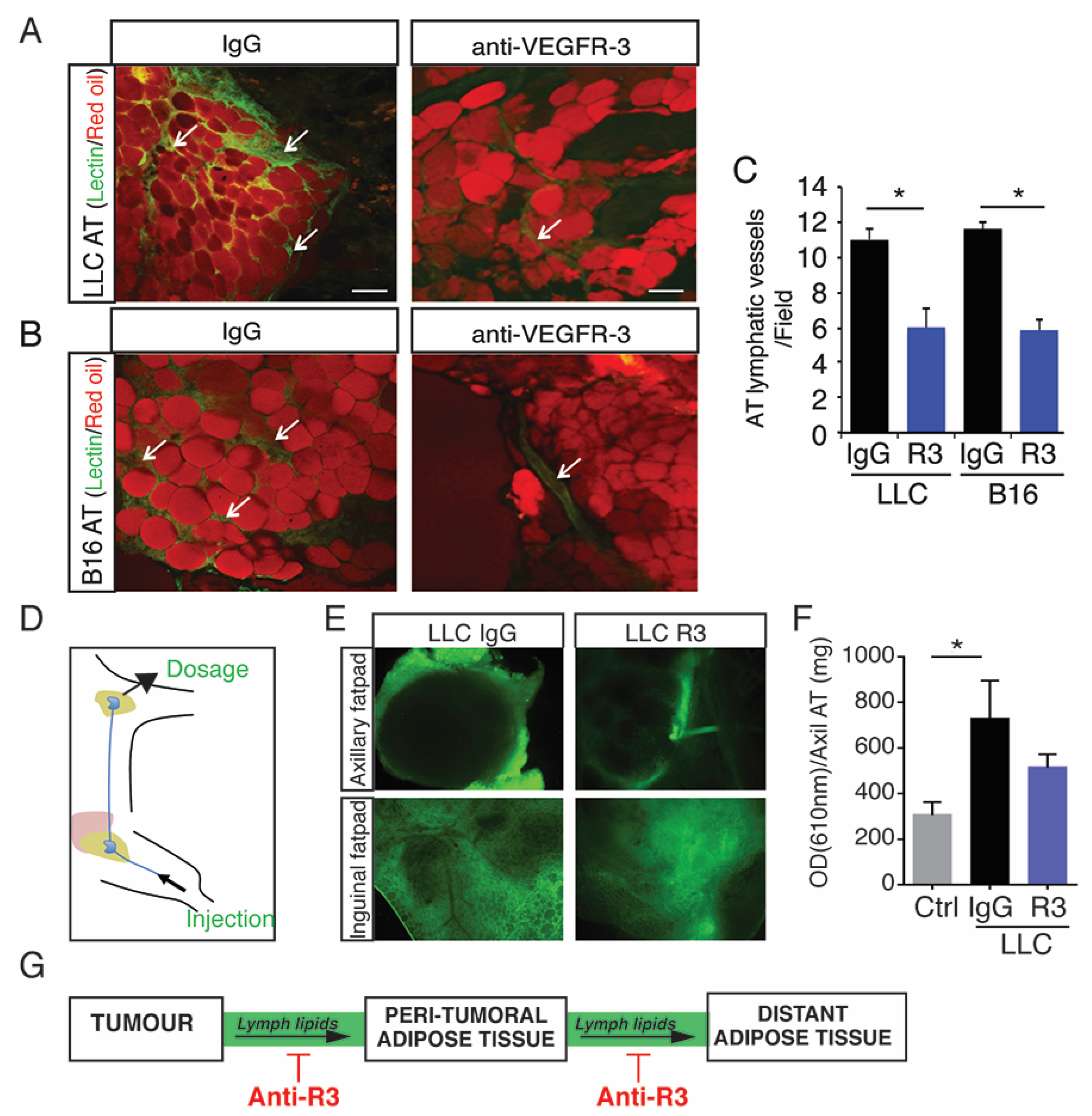
2.3. Blocking Peritumoral AT Lymphangiogenesis Decreases Lymph Circulating Lipid Levels
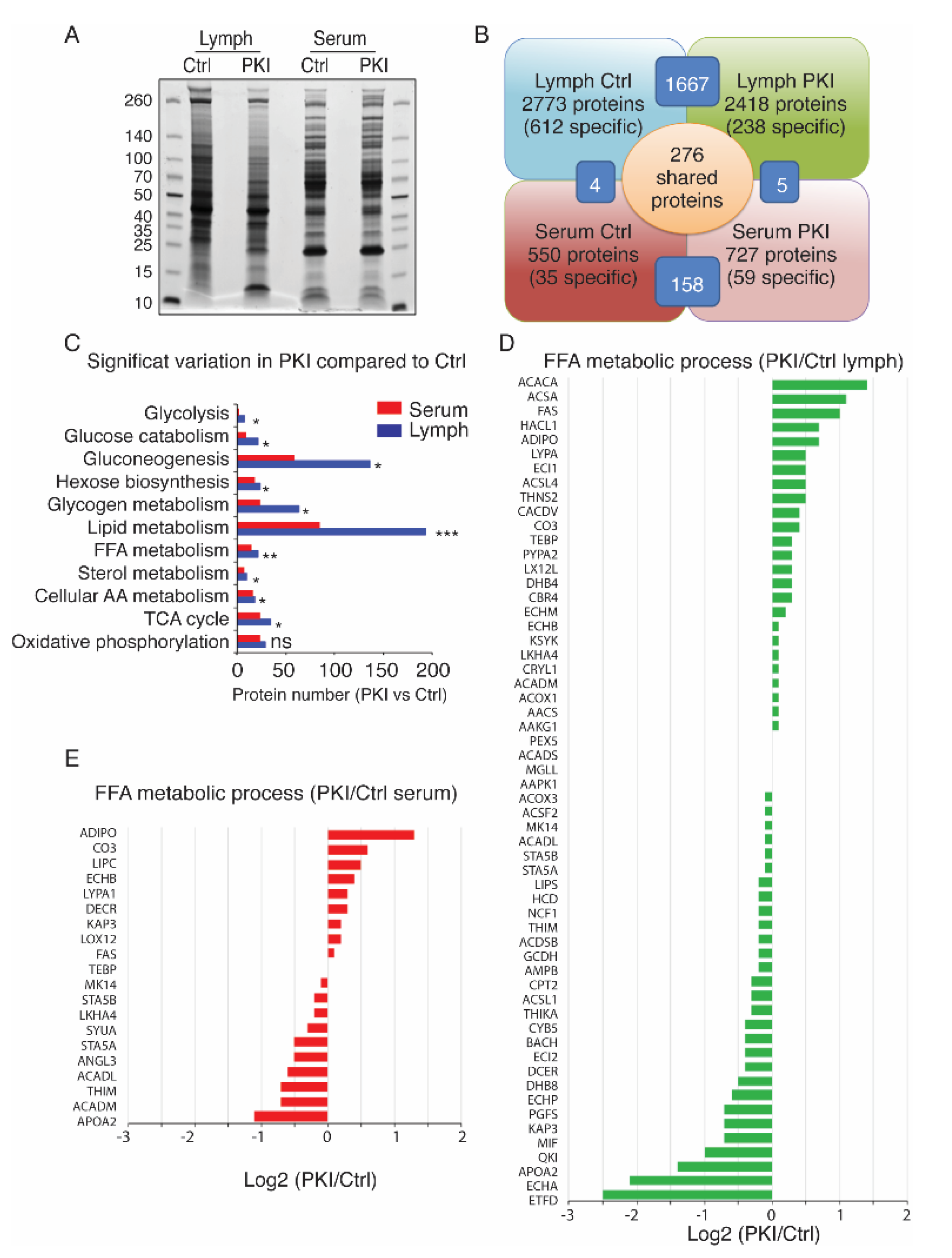
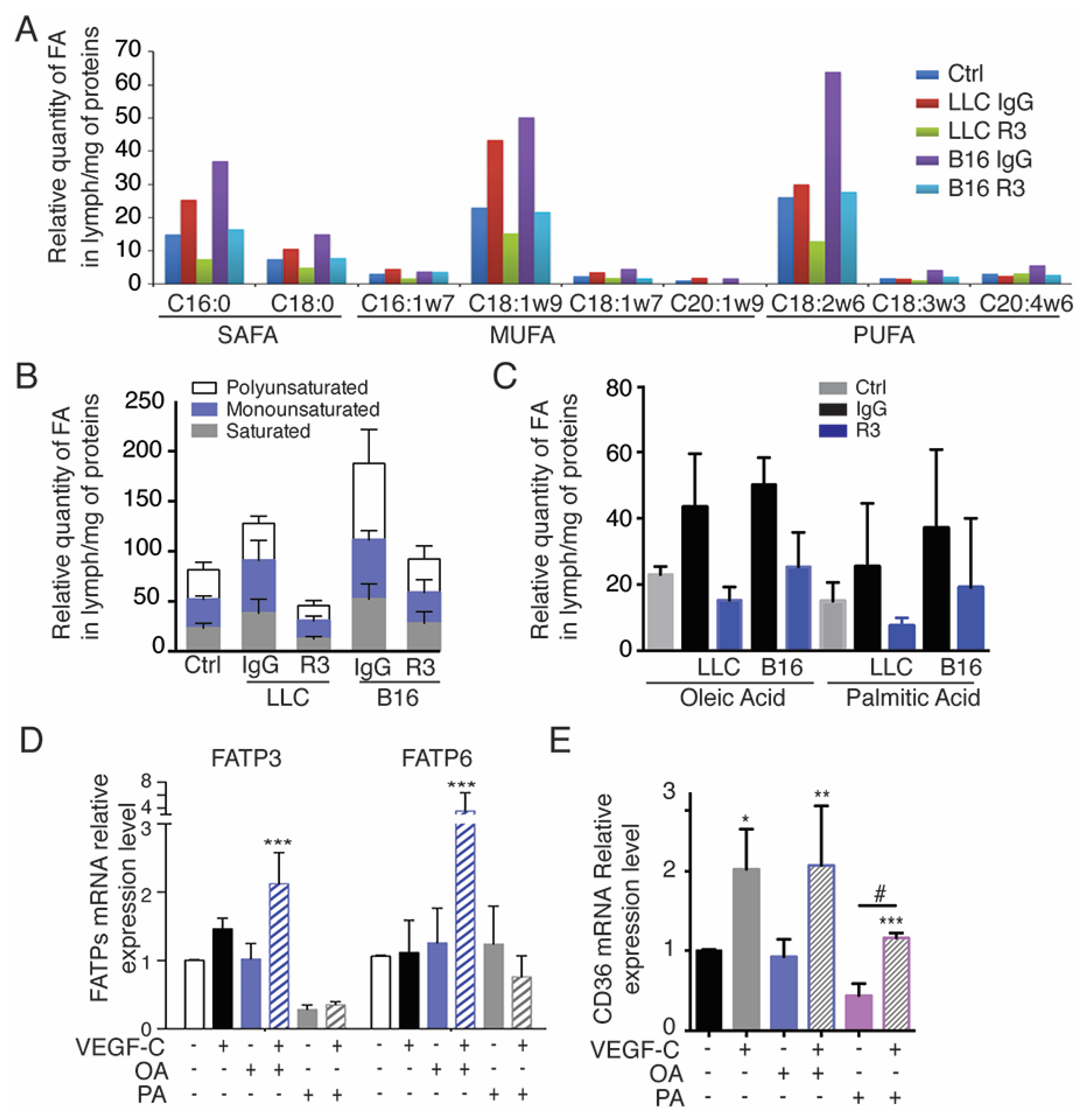
2.4. Tumor-Bearing Mice Exhibit Increased Level of Circulating Free Fatty Acids in Lymph
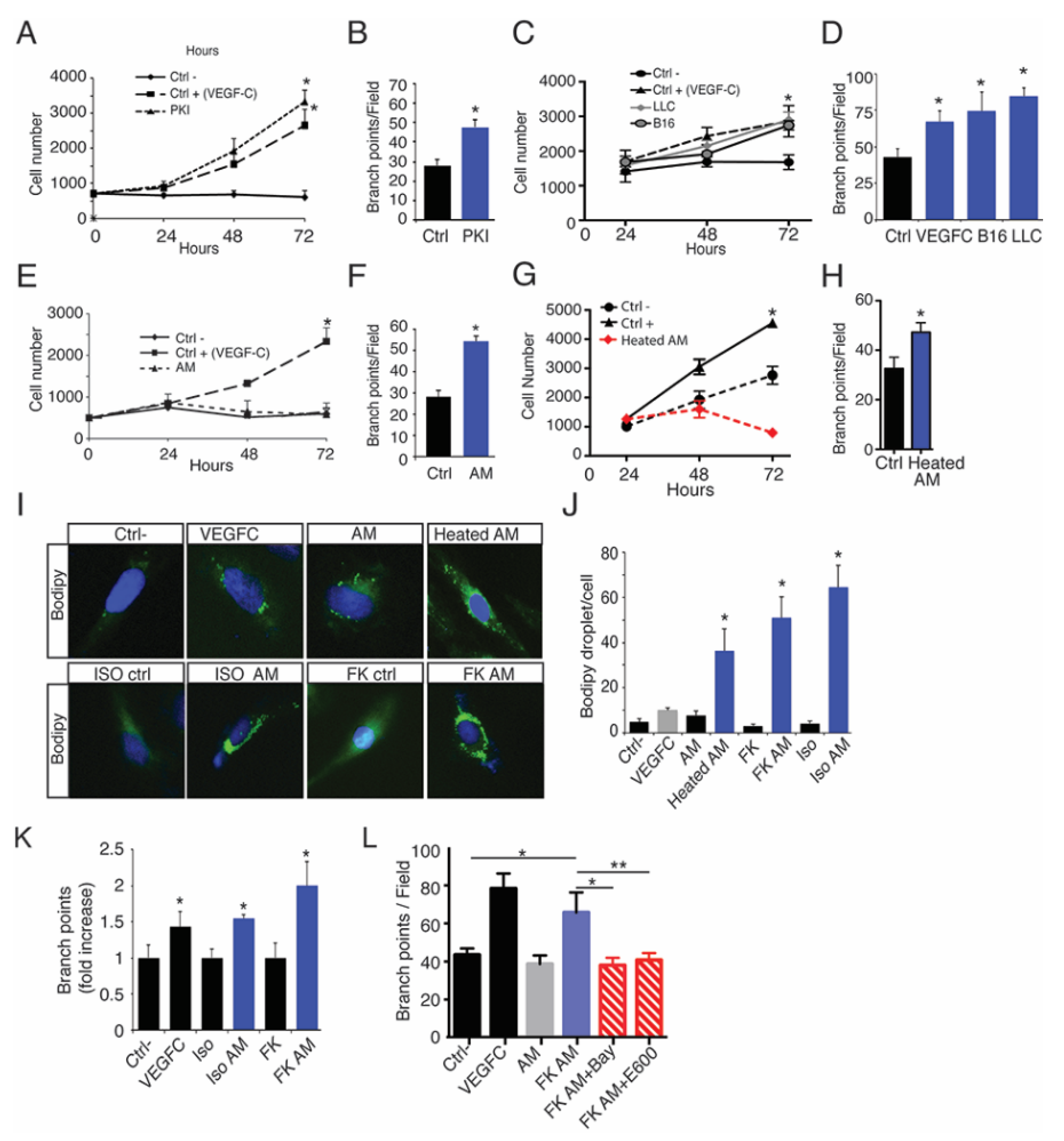
2.5. Oleic Acid Released by Adipose Tissue Drive LEC Sprouting
2.6. Lymphatic FFAs Stimulate LEC Function
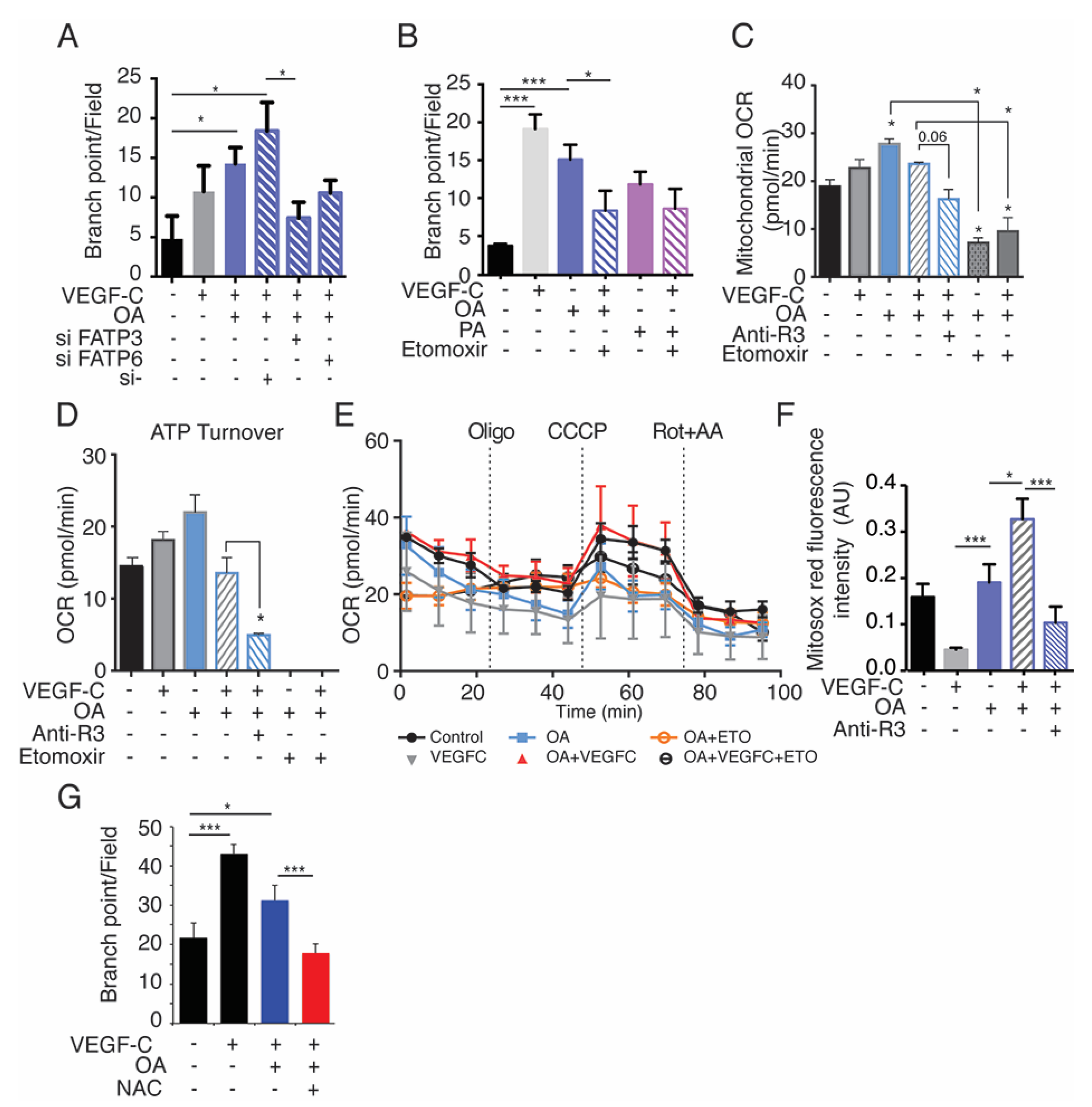
2.7. Exogenous Oleic Acid Stimulates LEC Beta-Oxidation
3. Discussion
4. Material and Methods
4.1. Human Tissues
4.2. Animal Study
4.3. Cell Culture
4.4. Chemical and Reagents
4.5. Immunohistochemistry
4.6. Labeling of the Lymphatic Compartment in AT
4.7. In Vivo Treatment with Anti-VEGFR2 and VEGFR3 Blocking Antibodies
4.8. Lymph Collection
4.9. ProteoMiner Protein Equalization and Analysis of Peptides by LC-MS/MS
4.10. Free Fatty Acid Methyl Ester (FAME) Analysis
4.11. Sprouting Assay
4.12. Heated Adipocyte Medium
4.13. Bodipy and Red Oil Uptake Assay
4.14. Quantitative Real-Time RT-PCR
4.15. MitoSOX Red
4.16. Seahorse Assays
4.17. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.G.; Lytle, N.K.; Jaquish, D.V.; Park, F.D.; Ito, T.; Bajaj, J.; Koechlein, C.S.; Zimdahl, B.; Yano, M.; Kopp, J.L.; et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 2016, 534, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [Green Version]
- Skobe, M.; Hamberg, L.M.; Hawighorst, T.; Schirner, M.; Wolf, G.L.; Alitalo, K.; Detmar, M. Concurrent Induction of Lymphangiogenesis, Angiogenesis, and Macrophage Recruitment by Vascular Endothelial Growth Factor-C in Melanoma. Am. J. Pathol. 2001, 159, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Morfoisse, F.; Tatin, F.; Hantelys, F.; Adoue, A.; Helfer, A.-C.; Cassant-Sourdy, S.; Pujol, F.; Gomez-Brouchet, A.; Ligat, L.; Lopez, F.; et al. Nucleolin Promotes Heat Shock–Associated Translation of VEGF-D to Promote Tumor Lymphangiogenesis. Cancer Res. 2016, 76, 4394–4405. [Google Scholar] [CrossRef] [Green Version]
- Tatin, F.; Renaud-Gabardos, E.; Godet, A.-C.; Hantelys, F.; Pujol, F.; Morfoisse, F.; Calise, D.; Viars, F.; Valet, P.; Masri, B.; et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Garmy-Susini, B.; Avraamides, C.J.; Desgrosellier, J.S.; Schmid, M.C.; Foubert, P.; Ellies, L.G. PI3Kalpha activates integrin al-pha4beta1 to establish a metastatic niche in lymph nodes. Proc. Natl. Acad. Sci. USA 2013, 110, 9042–9047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J. Clin. Investig. 2007, 117, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Chen, G.; You, T.; Yao, J.; Jiang, Q.; Lin, X.; Shen, X.; Qiao, Y.; Lin, L. High FFA-induced proliferation and apoptosis in human umbilical vein endothelial cell partly through Wnt/beta-catenin signal pathway. Mol. Cell Biochem. 2010, 338, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Investig. 1997, 100, 1230–1239. [Google Scholar] [CrossRef]
- Camargo, A.; Meneses, M.E.; Perez-Martinez, P.; Delgado-Lista, J.; Jimenez-Gomez, Y.; Cruz-Teno, C.; Tinahones, F.J.; Paniagua, J.A.; Perez-Jimenez, F.; Roche, H.M.; et al. Dietary fat differential-ly influences the lipids storage on the adipose tissue in metabolic syndrome patients. Eur. J. Nutr. 2014, 53, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Tvrzicka, E.; Kremmyda, L.-S.; Stankova, B.; Zak, A. Fatty acids AS biocompounds: Their role in human metabolism, health and disease—A review. Part 1: Classification, dietary sources and biological functions. Biomed. Pap. 2011, 155, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Tysseling, K.A.; Rice, J.; Pham, M.; Haji, L.; Gallis, B.M.; Baas, A.S.; Paramsothy, P.; Giachelli, C.M.; Corson, M.A.; et al. Free fatty acid impairment of nitric oxide production in endo-thelial cells is mediated by IKKbeta. Arter. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef] [PubMed]
- Djoussé, L.; Bartz, T.M.; Ix, J.H.; Kochar, J.; Kizer, J.R.; Gottdiener, J.S.; Tracy, R.P.; Mozaffarian, D.; Siscovick, D.S.; Mukamal, K.J.; et al. Fatty acid-binding protein 4 and incident heart failure: The Cardiovascular Health Study. Eur. J. Hear. Fail. 2013, 15, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Effects of Free Fatty Acids (FFA) on Glucose Metabolism: Significance for Insulin Resistance and Type 2 Diabetes. Exp. Clin. Endocrinol. Diabetes 2003, 111, 121–124. [Google Scholar] [CrossRef]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Broniarek, I.; Koziel, A.; Jarmuszkiewicz, W. The effect of chronic exposure to high palmitic acid concentrations on the aerobic metabolism of human endothelial EA.hy926 cells. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1541–1554. [Google Scholar] [CrossRef] [Green Version]
- Schwingshackl, L.; Christoph, M.; Hoffmann, G. Effects of Olive Oil on Markers of Inflammation and Endothelial Func-tion-A Systematic Review and Meta-Analysis. Nutrients 2015, 7, 7651–7675. [Google Scholar] [CrossRef] [Green Version]
- Sawane, M.; Kajiya, K.; Kidoya, H.; Takagi, M.; Muramatsu, F.; Takakura, N. Apelin inhibits diet-induced obesity by enhanc-ing lymphatic and blood vessel integrity. Diabetes 2013, 62, 1970–1980. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; HariKumar, S.; Negi, P.; Upadhyay, N.; Garg, R. Quetiapine Fumarate Loaded Nanostructured Lipid Carrier for Enhancing Oral Bioavailability: Design, Development and Pharmacokinetic Assessment. Curr. Drug Deliv. 2021, 18, 184–198. [Google Scholar] [CrossRef]
- Mishra, A.; Imam, S.S.; Aqil, M.; Ahad, A.; Sultana, Y.; Ameeduzzafar; Ali, A. Carvedilol nano lipid carriers: Formulation, characterization and in-vivo evaluation. Drug Deliv. 2016, 23, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Han, L.; He, J.; Lv, J.; Pan, R.; Lv, T. A high serum-free fatty acid level is associated with cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing mel-anoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; García-Caballero, M.; Missiaen, R.; Huang, H.; Brüning, U.; et al. The role of fatty acid beta-oxidation in lym-phangiogenesis. Nature 2016, 542, 49–54. [Google Scholar] [CrossRef]
- Olson, P.; Chu, G.C.; Perry, S.R.; Nolan-Stevaux, O.; Hanahan, D. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, E1275–E1284. [Google Scholar] [CrossRef] [Green Version]
- Wiig, H.; Keskin, D.; Kalluri, R. Interaction between the extracellular matrix and lymphatics: Consequences for lymphan-giogenesis and lymphatic function. Matrix Biol. 2010, 29, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose Triglyceride Lipase Contributes to Cancer-Associated Cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Grüner, B.M.; Schulze, C.J.; Yang, D.; Ogasawara, D.; Dix, M.M.; Rogers, Z.N.; Chuang, C.-H.; McFarland, C.D.; Chiou, S.-H.; Brown, J.M.; et al. An in vivo multiplexed small-molecule screening platform. Nat. Methods 2016, 13, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morfoisse, F.; Renaud, E.; Hantelys, F.; Prats, A.-C.; Garmy-Susini, B. Role of hypoxia and vascular endothelial growth factors in lymphangiogenesis. Mol. Cell. Oncol. 2014, 1, e29907. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Brown, N.F. The Mitochondrial Carnitine Palmitoyltransferase System—From Concept to Molecular Analysis. JBIC J. Biol. Inorg. Chem. 1997, 244, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.E.; Kang, Y.; Jang, H.J.; Shin, H.C.; Kim, H.E.; Kim, H.S.; Kim, H.J.; Kim, D.J.; Lee, K.W. Involvement of glycogen synthase kinase-3beta in palmitate-induced human umbilical vein endothelial cell apoptosis. J. Vasc. Res. 2007, 44, 365–374. [Google Scholar] [CrossRef]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10, eaag0945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Angiogenesis and Vascular Functions in Modulation of Obesity, Adipose Metabolism, and Insulin Sensitivity. Cell Metab. 2013, 18, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Barajas, S.; Lammoglia, G.M.; Reyna, A.J.; Morley, T.S.; Johnson, J.A.; Scherer, P.E.; Rutkowski, J.M. Vascular Endothelial Growth Factor–D (VEGF-D) Overexpression and Lymphatic Expansion in Murine Adipose Tissue Improves Metabolism in Obesity. Am. J. Pathol. 2019, 189, 924–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.C.; D’Alessandro, A.; Clement, C.C.; Santambrogio, L. Lymph formation, composition and circulation: A prote-omics perspective. Int. Immunol. 2015, 27, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endo-thelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimeno, R.E.; Ortegon, A.M.; Patel, S.; Punreddy, S.; Ge, P.; Sun, Y.; Lodish, H.F.; Stahl, A. Characterization of a Heart-specific Fatty Acid Transport Protein. J. Biol. Chem. 2003, 278, 16039–16044. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, R.; Hirata, K.; Kawashima, S.; Yokoyama, M. Unsaturated free fatty acids inhibit Ca2+ mobilization and NO release in endothelial cells. Kobe J. Med. Sci. 2001, 47, 211–219. [Google Scholar] [PubMed]
- Douin-Echinard, V.; Bornes, S.; Rochaix, P.; Tilkin, A.-F.; Péron, J.-M.; Bonnet, J.; Favre, G.; Couderc, B. The expression of CD70 and CD80 by gene-modified tumor cells induces an antitumor response depending on the MHC status. Cancer Gene Ther. 2000, 7, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernemalm, M.; Orre, L.M.; Lengqvist, J.; Wikström, P.; Lewensohn, R.; Lehtiö, J. Evaluation of Three Principally Different Intact Protein Prefractionation Methods for Plasma Biomarker Discovery. J. Proteome Res. 2008, 7, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Thulasiraman, V.; Lin, S.; Gheorghiu, L.; Lathrop, J.; Lomas, L.; Hammond, D.; Boschetti, E. Reduction of the concentration difference of proteins in biological liquids using a library of combinatorial ligands. Electrophoresis 2005, 26, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Lillington, J.M.; Trafford, D.J.; Makin, H.L. A rapid and simple method for the esterification of fatty acids and steroid car-boxylic acids prior to gas-liquid chromatography. Clin. Chim. Acta Int. J. Clin. Chem. 1981, 111, 91–98. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morfoisse, F.; De Toni, F.; Nigri, J.; Hosseini, M.; Zamora, A.; Tatin, F.; Pujol, F.; Sarry, J.-E.; Langin, D.; Lacazette, E.; et al. Coordinating Effect of VEGFC and Oleic Acid Participates to Tumor Lymphangiogenesis. Cancers 2021, 13, 2851. https://doi.org/10.3390/cancers13122851
Morfoisse F, De Toni F, Nigri J, Hosseini M, Zamora A, Tatin F, Pujol F, Sarry J-E, Langin D, Lacazette E, et al. Coordinating Effect of VEGFC and Oleic Acid Participates to Tumor Lymphangiogenesis. Cancers. 2021; 13(12):2851. https://doi.org/10.3390/cancers13122851
Chicago/Turabian StyleMorfoisse, Florent, Fabienne De Toni, Jeremy Nigri, Mohsen Hosseini, Audrey Zamora, Florence Tatin, Françoise Pujol, Jean-Emmanuel Sarry, Dominique Langin, Eric Lacazette, and et al. 2021. "Coordinating Effect of VEGFC and Oleic Acid Participates to Tumor Lymphangiogenesis" Cancers 13, no. 12: 2851. https://doi.org/10.3390/cancers13122851
APA StyleMorfoisse, F., De Toni, F., Nigri, J., Hosseini, M., Zamora, A., Tatin, F., Pujol, F., Sarry, J. -E., Langin, D., Lacazette, E., Prats, A. -C., Tomasini, R., Galitzky, J., Bouloumié, A., & Garmy-Susini, B. (2021). Coordinating Effect of VEGFC and Oleic Acid Participates to Tumor Lymphangiogenesis. Cancers, 13(12), 2851. https://doi.org/10.3390/cancers13122851