
Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer

 ,
, 
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Clinical Features of Breast Cancer
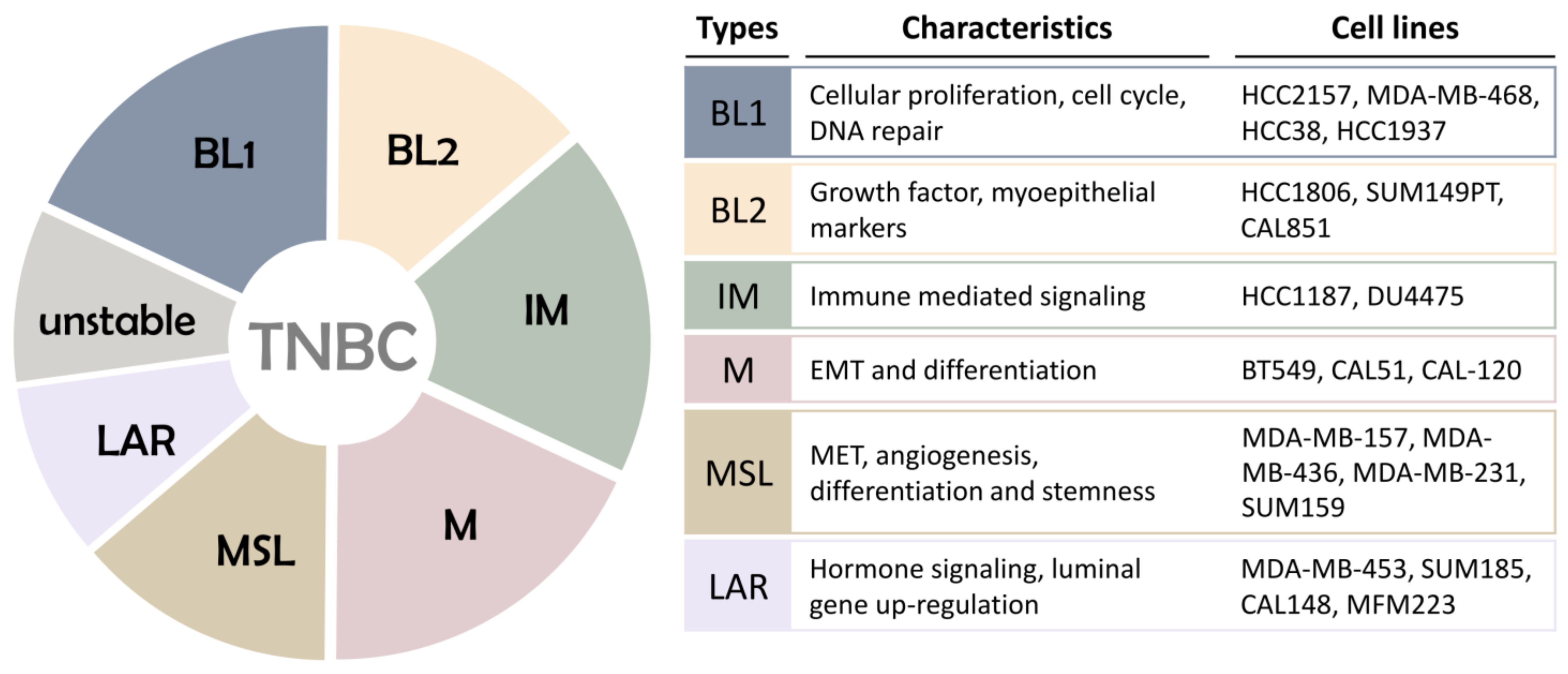
3. Molecular Types of Triple Negative Breast Cancer
4. Prognostic Biomarkers of TNBC
4.1. BRCA 1/2
4.2. ALDH1A1
4.3. CXCR4
4.4. p16INK4a
4.5. ATM
4.6. PTEN
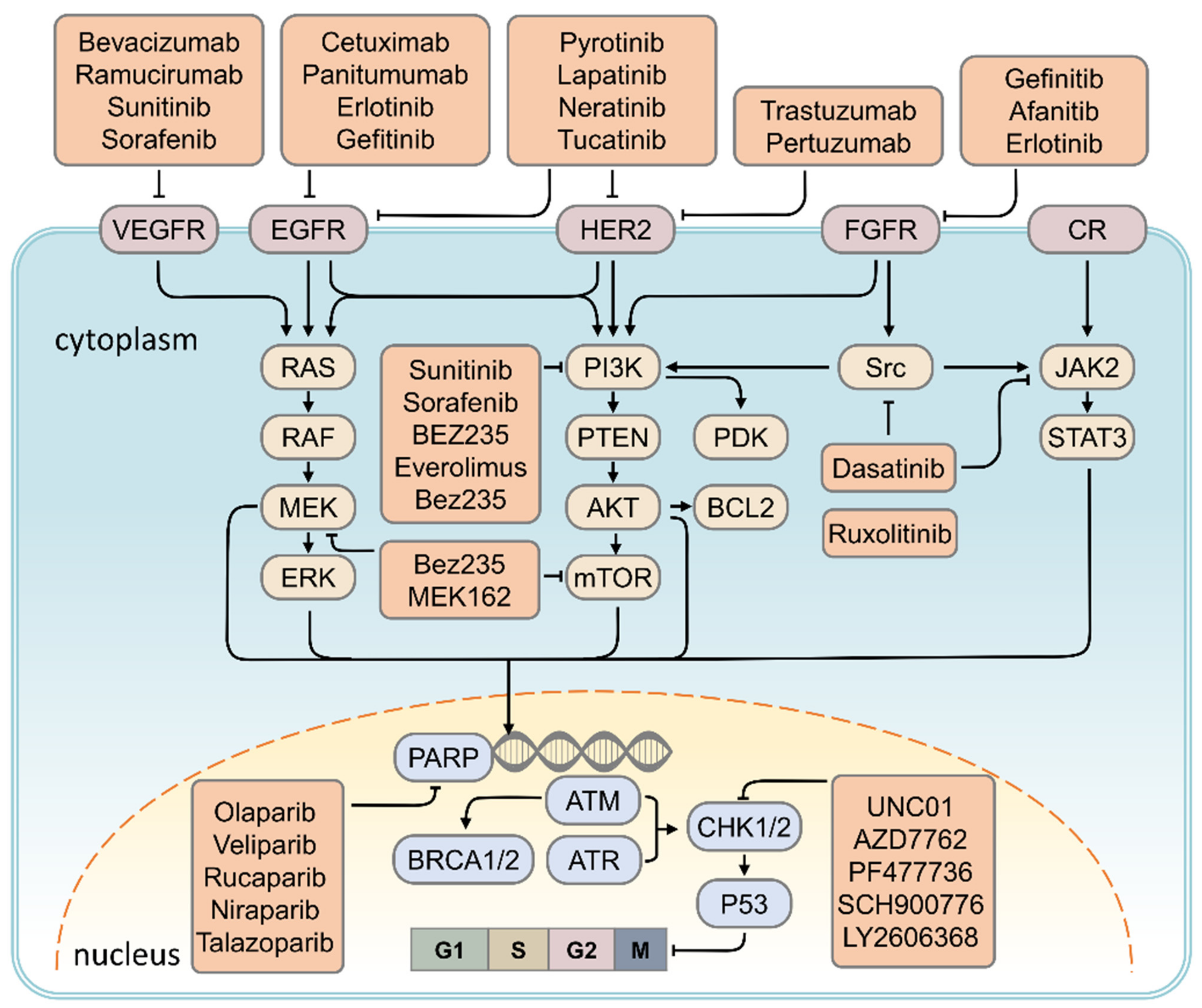
5. Therapeutic Strategy for TNBC
5.1. HER2 Inhibitor
Targeting RTK Signaling
5.2. VEGF Inhibitor
5.3. PARP Inhibitor
5.4. CHK1 Inhibitor
5.5. EGFR Inhibitor
5.6. Targeting Based on Cell Proliferation and Survival-Dependent Pathways in TNBC
5.6.1. Targeting of the PI3K/AKT/mTOR Signaling Pathway
5.6.2. Targeting of MAPK Signaling Pathway
5.6.3. Targeting the JAK2/STAT3 Signaling Pathway
5.6.4. Targeting of the Src Signaling Pathway
6. Immune Checkpoint Blockade
7. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Toriola, A.T.; Colditz, G.A. Trends in breast cancer incidence and mortality in the United States: Implications for prevention. Breast Cancer Res. Treat. 2013, 138, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ellison, R.C.; Zhang, Y.; McLennan, C.E.; Rothman, K.J. Exploring the relation of alcohol consumption to risk of breast cancer. Am. J. Epidemiol. 2001, 154, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, K.; Mao, K.; Su, F.; Liu, Q.; Jacobs, L.K. The prognostic value of age for invasive lobular breast cancer depending on estrogen receptor and progesterone receptor-defined subtypes: A NCDB analysis. Oncotarget 2016, 7, 6063–6073. [Google Scholar] [CrossRef] [Green Version]
- Stavros, A.T.; Thickman, D.; Rapp, C.L.; Dennis, M.A.; Parker, S.H.; Sisney, G.A. Solid breast nodules: Use of sonography to distinguish between benign and malignant lesions. Radiology 1995, 196, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Flobbe, K.; Bosch, A.; Kessels, A.H. The additional diagnostic value of ultrasonography in the diagnosis of breast cancer. Arch. Intern. Med. 2003, 163, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishna, S.; Agarwal, S.; Parikh, P.M.; Kaur, K.; Panwar, S.; Sharma, S.; Dey, A.; Saxena, K.K.; Chandra, M.; Sud, S. Role of magnetic resonance imaging in breast cancer management. South Asian J. Cancer 2018, 7, 69–71. [Google Scholar] [CrossRef]
- Bartlett, J.M.; Brookes, C.L.; Robson, T.; van de Velde, C.J.; Billingham, L.J.; Campbell, F.M.; Grant, M.; Hasenburg, A.; Hille, E.T.; Kay, C.; et al. Estrogen receptor and progesterone receptor as predictive biomarkers of response to endocrine therapy: A prospectively powered pathology study in the Tamoxifen and Exemestane Adjuvant Multinational trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1531–1538. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Chu, P.Y.; Yiang, G.T.; Wu, M.Y. The Molecular Mechanism of Epithelial-Mesenchymal Transition for Breast Carcinogenesis. Biomolecules 2019, 9, 476. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Trotta, L.; Norberg, A.; Taskinen, M.; Beziat, V.; Degerman, S.; Wartiovaara-Kautto, U.; Valimaa, H.; Jahnukainen, K.; Casanova, J.L.; Seppanen, M.; et al. Diagnostics of rare disorders: Whole-exome sequencing deciphering locus heterogeneity in telomere biology disorders. Orphanet J. Rare Dis. 2018, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- Gerratana, L.; Basile, D.; Buono, G.; de Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; de Santo, I.; del Mastro, L.; et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1 and BRCA2—Update and implications on the genetics of breast cancer: A clinical perspective. Clin. Genet. 2014, 85, 1–4. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Wang, Z.; Liu, M.; Qi, Z.; Meng, J.; Sun, J.; Yang, G. Aurora-A: A potential DNA repair modulator. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 2831–2836. [Google Scholar] [CrossRef]
- Park, J.Y.; Zhang, F.; Andreassen, P.R. PALB2: The hub of a network of tumor suppressors involved in DNA damage responses. Biochim. Biophys. Acta 2014, 1846, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, L.; Panchal, S.; Goodwin, P. Prognosis of BRCA-associated breast cancer: A summary of evidence. Breast Cancer Res. Treat. 2010, 119, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, A.J.; Schmidt, M.K.; van’t Veer, L.J.; Tollenaar, R.A.; van Leeuwen, F.E. Worse breast cancer prognosis of BRCA1/BRCA2 mutation carriers: What’s the evidence? A systematic review with meta-analysis. PLoS ONE 2015, 10, e0120189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, D.L.; Duan, J.J.; Xu, S.L.; Zhang, J.F.; Yang, X.J.; Zhang, X.; Cui, Y.H.; Bian, X.W.; Yu, S.C. ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 2014, 14, 444. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.Y.; Thike, A.A.; Tan, P.H. ALDH1 expression is enriched in breast cancers arising in young women but does not predict outcome. Br. J. Cancer 2013, 109, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Bi, L.R.; Xu, N.; Yang, H.M.; Zhang, H.T.; Ding, Y.; Shi, A.P.; Fan, Z.M. The expression of aldehyde dehydrogenase 1 in invasive primary breast tumors and axillary lymph node metastases is associated with poor clinical prognosis. Pathol. Res. Pract. 2013, 209, 555–561. [Google Scholar] [CrossRef]
- Yoshioka, T.; Umekita, Y.; Ohi, Y.; Souda, M.; Sagara, Y.; Rai, Y.; Tanimoto, A. Aldehyde dehydrogenase 1 expression is a predictor of poor prognosis in node-positive breast cancers: A long-term follow-up study. Histopathology 2011, 58, 608–616. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, e6561. [Google Scholar] [CrossRef]
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009, 100, 1062–1068. [Google Scholar] [CrossRef]
- Resetkova, E.; Reis-Filho, J.S.; Jain, R.K.; Mehta, R.; Thorat, M.A.; Nakshatri, H.; Badve, S. Prognostic impact of ALDH1 in breast cancer: A story of stem cells and tumor microenvironment. Breast Cancer Res. Treat. 2010, 123, 97–108. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D. CXCR4 chemokine receptor overview: Biology, pathology and applications in imaging and therapy. Theranostics 2013, 3, 1–2. [Google Scholar] [CrossRef]
- Xu, T.P.; Shen, H.; Liu, L.X.; Shu, Y.Q. The impact of chemokine receptor CXCR4 on breast cancer prognosis: A meta-analysis. Cancer Epidemiol. 2013, 37, 725–731. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.D.; Panu, L.; Holm, N.T.; Li, B.D.; Johnson, L.W.; Zhang, S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J. Surg. Res. 2010, 159, 689–695. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Liu, G.; Zhu, X.; Chen, Y. High level of CXCR4 in triple-negative breast cancer specimens associated with a poor clinical outcome. Acta Med. Okayama 2013, 67, 369–375. [Google Scholar]
- Parker, C.C.; Kim, R.H.; Li, B.D.; Chu, Q.D. The chemokine receptor CXCR4 as a novel independent prognostic marker for node-positive breast cancer patients. J. Surg. Oncol. 2012, 106, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Jung, W.H.; Koo, J.S. Expression of p16 and pRB in invasive breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8209–8217. [Google Scholar]
- Wang, L.; Tang, L.; Xie, R.; Nie, W.; Chen, L.; Guan, X. p16 promoter hypermethylation is associated with increased breast cancer risk. Mol. Med. Rep. 2012, 6, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.W.; Sohn, J.H.; Kim, D.H.; Choi, Y.J.; Park, Y.L.; Kim, K.; Cho, Y.H.; Pyo, J.S.; Kim, J.H. Overexpressions of Cyclin B1, cdc2, p16 and p53 in human breast cancer: The clinicopathologic correlations and prognostic implications. Yonsei Med. J. 2011, 52, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Vallian, S.; Sedaghat, M.; Nassiri, I.; Frazmand, A. Methylation status of p16 INK4A tumor suppressor gene in Iranian patients with sporadic breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 991–996. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Lee, J.J.; Ko, E.; Cho, J.; Park, H.Y.; Lee, J.E.; Nam, S.J.; Kim, D.H.; Cho, E.Y. Methylation and Immunoexpression of p16(INK4a) Tumor Suppressor Gene in Primary Breast Cancer Tissue and Their Quantitative p16(INK4a) Hypermethylation in Plasma by Real-Time PCR. Korean J. Pathol. 2012, 46, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, X.; Zhang, J. Inhibition of Breast Tumor Cell Growth by Ectopic Expression of p16/INK4A via Combined Effects of Cell Cycle Arrest, Senescence and Apoptotic Induction, and Angiogenesis Inhibition. J. Cancer 2012, 3, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subhawong, A.P.; Subhawong, T.; Nassar, H.; Kouprina, N.; Begum, S.; Vang, R.; Westra, W.H.; Argani, P. Most basal-like breast carcinomas demonstrate the same Rb−/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am. J. Surg. Pathol. 2009, 33, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sugianto, J.; Sarode, V.; Peng, Y. Ki-67 expression is increased in p16-expressing triple-negative breast carcinoma and correlates with p16 only in p53-negative tumors. Hum. Pathol. 2014, 45, 802–809. [Google Scholar] [CrossRef]
- Karray-Chouayekh, S.; Baccouche, S.; Khabir, A.; Sellami-Boudawara, T.; Daoud, J.; Frikha, M.; Jlidi, R.; Gargouri, A.; Mokdad-Gargouri, R. Prognostic significance of p16INK4a/p53 in Tunisian patients with breast carcinoma. Acta Histochem. 2011, 113, 508–513. [Google Scholar] [CrossRef]
- Arima, Y.; Hayashi, N.; Hayashi, H.; Sasaki, M.; Kai, K.; Sugihara, E.; Abe, E.; Yoshida, A.; Mikami, S.; Nakamura, S.; et al. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int. J. Cancer 2012, 130, 2568–2579. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Reitnauer, P.J.; Morrell, D.; Chase, C.L. Breast and other cancers in families with ataxia-telangiectasia. N. Engl. J. Med. 1987, 316, 1289–1294. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Wooster, R.; Weber, B.L. Breast cancer genetics: What we know and what we need. Nat. Med. 2001, 7, 552–556. [Google Scholar] [CrossRef]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef]
- Rondeau, S.; Vacher, S.; De Koning, L.; Briaux, A.; Schnitzler, A.; Chemlali, W.; Callens, C.; Lidereau, R.; Bieche, I. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br. J. Cancer 2015, 112, 1059–1066. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkas, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Domagala, P.; Jakubowska, A.; Jaworska-Bieniek, K.; Kaczmarek, K.; Durda, K.; Kurlapska, A.; Cybulski, C.; Lubinski, J. Prevalence of Germline Mutations in Genes Engaged in DNA Damage Repair by Homologous Recombination in Patients with Triple-Negative and Hereditary Non-Triple-Negative Breast Cancers. PLoS ONE 2015, 10, e0130393. [Google Scholar] [CrossRef]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Kuo, W.H.; Huang, A.C.; Lu, Y.S.; Lin, C.H.; Kuo, S.H.; Wang, M.Y.; Liu, C.Y.; Cheng, F.T.; Yeh, M.H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [Green Version]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; Tsao, M.S.; Macpherson, D.; Suzuki, A.; Chapman, W.B.; Mak, T.W. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 2000, 60, 3605–3611. [Google Scholar]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z. Loss of PTEN expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, E.; Oki, E.; Kimura, Y.; Yamanaka, T.; Egashira, A.; Nishida, K.; Koga, T.; Morita, M.; Kakeji, Y.; Maehara, Y. Coexistence of the loss of heterozygosity at the PTEN locus and HER2 overexpression enhances the Akt activity thus leading to a negative progesterone receptor expression in breast carcinoma. Breast Cancer Res. Treat. 2007, 101, 249–257. [Google Scholar] [CrossRef]
- Yndestad, S.; Austreid, E.; Knappskog, S.; Chrisanthar, R.; Lilleng, P.K.; Lonning, P.E.; Eikesdal, H.P. High PTEN gene expression is a negative prognostic marker in human primary breast cancers with preserved p53 function. Breast Cancer Res. Treat. 2017, 163, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.C.; Reis-Filho, J.S.; Lakhani, S.R. Phenotype-genotype correlation in familial breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 27–40. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.; Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Anton, A.; Lluch, A.; et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. J. Clin. Oncol. 2005, 23, 4265–4274. [Google Scholar] [CrossRef]
- Chick, R.C.; Clifton, G.T.; Hale, D.F.; Vreeland, T.J.; Hickerson, A.T.; Kemp Bohan, P.M.; McCarthy, P.M.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; et al. Subgroup analysis of nelipepimut-S plus GM-CSF combined with trastuzumab versus trastuzumab alone to prevent recurrences in patients with high-risk, HER2 low-expressing breast cancer. Clin. Immunol. 2021, 225, 108679. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab versus Trastuzumab to Prevent Recurrences in Patients with High-Risk HER2 Low-Expressing Breast Cancer. Clin. Cancer Res. 2020, 26, 2515–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [PubMed]
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 2016, 27, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Toh, U.; Tanaka, M.; Saimura, M.; Okumura, Y.; Saito, T.; Tanaka, T.; Teraoka, M.; Shimada, K.; Katayama, K.; et al. Role of HER2-Related Biomarkers (HER2, p95HER2, HER3, PTEN, and PIK3CA) in the Efficacy of Lapatinib plus Capecitabine in HER2-Positive Advanced Breast Cancer Refractory to Trastuzumab. Oncology 2017, 93, 51–61. [Google Scholar] [CrossRef]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Copeland, A.C.; Anders, C.K. Dual HER2-Targeting in the Adjuvant Setting: Where We Have Been and Where We Are Going. Oncology 2018, 32, 483–487. [Google Scholar]
- Deeks, E.D. Neratinib: First Global Approval. Drugs 2017, 77, 1695–1704. [Google Scholar] [CrossRef]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer 2020, 19, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Chellappan, D.K.; Chellian, J.; Ng, Z.Y.; Sim, Y.J.; Theng, C.W.; Ling, J.; Wong, M.; Foo, J.H.; Yang, G.J.; Hang, L.Y.; et al. The role of pazopanib on tumour angiogenesis and in the management of cancers: A review. Biomed. Pharm. 2017, 96, 768–781. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Ruggieri, S.; Tamma, R.; Simone, G.; Mangia, A. Angiogenesis and Antiangiogenesis in Triple-Negative Breast cancer. Transl. Oncol. 2016, 9, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Labanca, V.; Bertolini, F. A Combinatorial Investigation of the Response to Anti-angiogenic Therapy in Breast Cancer: New Strategies for Patient Selection and Opportunities for Reconsidering Anti-VEGF, Anti-PI3K and Checkpoint Inhibition. EBioMedicine 2016, 10, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Lobo, C.; Lopes, G.; Baez, O.; Castrellon, A.; Ferrell, A.; Higgins, C.; Hurley, E.; Hurley, J.; Reis, I.; Richman, S.; et al. Final results of a phase II study of nab-paclitaxel, bevacizumab, and gemcitabine as first-line therapy for patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2010, 123, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.; Valero, V.; Tiangco, B.; Dakhil, S.; Brize, A.; Rugo, H.S.; Rivera, R.; Duenne, A.; Bousfoul, N.; Yardley, D.A. Second-line bevacizumab-containing therapy in patients with triple-negative breast cancer: Subgroup analysis of the RIBBON-2 trial. Breast Cancer Res. Treat. 2012, 133, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Liposits, G.; Loh, K.P.; Soto-Perez-de-Celis, E.; Dumas, L.; Battisti, N.M.L.; Kadambi, S.; Baldini, C.; Banerjee, S.; Lichtman, S.M. PARP inhibitors in older patients with ovarian and breast cancer: Young International Society of Geriatric Oncology review paper. J. Geriatr. Oncol. 2019, 10, 337–345. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef]
- Johnson, N.; Shapiro, G.I. Cyclin-dependent kinases (cdks) and the DNA damage response: Rationale for cdk inhibitor-chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin. Targets 2010, 14, 1199–1212. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Han, H.S.; Dieras, V.; Robson, M.; Palacova, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Seong, R.K.; Choi, Y.K.; Shin, O.S. MDA7/IL-24 is an anti-viral factor that inhibits influenza virus replication. J. Microbiol. 2016, 54, 695–700. [Google Scholar] [CrossRef]
- Dent, R.A.; Lindeman, G.J.; Clemons, M.; Wildiers, H.; Chan, A.; McCarthy, N.J.; Singer, C.F.; Lowe, E.S.; Watkins, C.L.; Carmichael, J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013, 15, R88. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.M.; Ansari, J.A.K.; El-Aziz, N.M.A.; Abozeed, W.N.; Warith, A.M.A.; Alsaleh, K.; Nabholtz, J.M. Triple Negative Breast Cancer: A Tale of Two Decades. Anticancer Agents Med. Chem. 2017, 17, 491–499. [Google Scholar] [CrossRef]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Huober, J.; Fasching, P.A.; Hanusch, C.; Rezai, M.; Eidtmann, H.; Kittel, K.; Hilfrich, J.; Schwedler, K.; Blohmer, J.U.; Tesch, H.; et al. Neoadjuvant chemotherapy with paclitaxel and everolimus in breast cancer patients with non-responsive tumours to epirubicin/cyclophosphamide (EC) ± bevacizumab—Results of the randomised GeparQuinto study (GBG 44). Eur. J. Cancer 2013, 49, 2284–2293. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Akcakanat, A.; Liu, S.; Green, M.C.; Murray, J.L.; Chen, H.; Palla, S.L.; Koenig, K.B.; Brewster, A.M.; Valero, V.; et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann. Oncol. 2014, 25, 1122–1127. [Google Scholar] [CrossRef]
- Rodon, J.; Perez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharm. 2018, 82, 285–298. [Google Scholar] [CrossRef]
- Patani, N.; Douglas-Jones, A.; Mansel, R.; Jiang, W.; Mokbel, K. Tumour suppressor function of MDA-7/IL-24 in human breast cancer. Cancer Cell Int. 2010, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.C.; Gadi, V. Dasatinib in breast cancer: Src-ing for response in all the wrong kinases. Ann. Transl. Med. 2018, 6 (Suppl. S1), S60. [Google Scholar] [CrossRef]
- Morris, P.G.; Rota, S.; Cadoo, K.; Zamora, S.; Patil, S.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; et al. Phase II Study of Paclitaxel and Dasatinib in Metastatic Breast Cancer. Clin. Breast Cancer 2018, 18, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef]
- Moulder, S.L.; Borges, V.F.; Baetz, T.; McSpadden, T.; Fernetich, G.; Murthy, R.K.; Chavira, R.; Guthrie, K.; Barrett, E.; Chia, S.K. Phase I Study of ONT-380, a HER2 Inhibitor, in Patients with HER2+—Advanced Solid Tumors, with an Expansion Cohort in HER2+ Metastatic Breast Cancer (MBC). Clin. Cancer Res. 2017, 23, 3529–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escriva-de-Romani, S.; Arumi, M.; Bellet, M.; Saura, C. HER2-positive breast cancer: Current and new therapeutic strategies. Breast 2018, 39, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Lee, K.W.; Oh, D.Y.; Lee, J.S.; Im, S.A.; Kim, D.W.; Han, S.W.; Kim, Y.J.; Kim, T.Y.; Kim, J.H.; et al. Phase 1 Studies of Poziotinib, an Irreversible Pan-HER Tyrosine Kinase Inhibitor in Patients with Advanced Solid Tumors. Cancer Res. Treat. 2018, 50, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsdorf, M.; Ingvar, C.; Jorgensen, L.; Tuxen, M.K.; Jakobsen, E.H.; Saetersdal, A.; Kimper-Karl, M.L.; Kroman, N.; Balslev, E.; Ejlertsen, B. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res. Treat. 2011, 126, 463–470. [Google Scholar] [CrossRef]
- Schuler, M.; Awada, A.; Harter, P.; Canon, J.L.; Possinger, K.; Schmidt, M.; de Greve, J.; Neven, P.; Dirix, L.; Jonat, W.; et al. A phase II trial to assess efficacy and safety of afatinib in extensively pretreated patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Layman, R.M.; Ruppert, A.S.; Lynn, M.; Mrozek, E.; Ramaswamy, B.; Lustberg, M.B.; Wesolowski, R.; Ottman, S.; Carothers, S.; Bingman, A.; et al. Severe and prolonged lymphopenia observed in patients treated with bendamustine and erlotinib for metastatic triple negative breast cancer. Cancer Chemother. Pharm. 2013, 71, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Meattini, I.; Livi, L.; Saieva, C.; Franceschini, D.; Scotti, V.; Mangoni, M.; Loi, M.; di Brina, L.; Zei, G.; Bonomo, P.; et al. Prognostic role of human epidermal growth factor receptor 2 status in premenopausal early breast cancer treated with adjuvant tamoxifen. Clin. Breast Cancer 2013, 13, 247–253. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting Epidermal Growth Factor Receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Basho, R.K.; Gilcrease, M.; Murthy, R.K.; Helgason, T.; Karp, D.D.; Meric-Bernstam, F.; Hess, K.R.; Herbrich, S.M.; Valero, V.; Albarracin, C.; et al. Targeting the PI3K/AKT/mTOR Pathway for the Treatment of Mesenchymal Triple-Negative Breast Cancer: Evidence from a Phase 1 Trial of mTOR Inhibition in Combination with Liposomal Doxorubicin and Bevacizumab. JAMA Oncol. 2017, 3, 509–515. [Google Scholar] [CrossRef]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel with or without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Guo, Z.; Luo, J.; Ellis, M.J.; Ma, C.X. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol. Cancer 2013, 12, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Chia, K.M.; Liu, J. Synergy between inhibitors of androgen receptor and MEK has therapeutic implications in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011, 13, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illiano, M.; Sapio, L.; Salzillo, A.; Capasso, L.; Caiafa, I.; Chiosi, E.; Spina, A.; Naviglio, S. Forskolin improves sensitivity to doxorubicin of triple negative breast cancer cells via Protein Kinase A-mediated ERK1/2 inhibition. Biochem. Pharm. 2018, 152, 104–113. [Google Scholar] [CrossRef]
- Qin, H.; Liu, X.; Li, F.; Miao, L.; Li, T.; Xu, B.; An, X.; Muth, A.; Thompson, P.R.; Coonrod, S.A.; et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. 2017, 409, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Luo, Q.; Xiao, Y.; Li, H.; Kong, L.; Ren, G. The implication from RAS/RAF/ERK signaling pathway increased activation in epirubicin treated triple negative breast cancer. Oncotarget 2017, 8, 108249–108260. [Google Scholar] [CrossRef] [Green Version]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Barni, S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 223–232. [Google Scholar] [CrossRef]
- Montero, J.C.; Esparis-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocana, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Haga, Y.; Higashisaka, K.; Yang, L.; Sekine, N.; Lin, Y.; Tsujino, H.; Nagano, K.; Tsutsumi, Y. Inhibition of Akt/mTOR pathway overcomes intrinsic resistance to dasatinib in triple-negative breast cancer. Biochem. Biophys. Res. Commun. 2020, 533, 672–678. [Google Scholar] [CrossRef]
- Kim, E.M.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J. Surg. Res. 2013, 185, 231–239. [Google Scholar] [CrossRef]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef]
- Li, C.J.; Chen, H.M.; Lai, J.C. Diagnostic, Prognostic, and Predictive Biomarkers in Breast Cancer. J. Oncol. 2020, 2020, 1835691. [Google Scholar] [CrossRef]
- Marinelli, D.; Mazzotta, M.; Pizzuti, L.; Krasniqi, E.; Gamucci, T.; Natoli, C.; Grassadonia, A.; Tinari, N.; Tomao, S.; Sperduti, I.; et al. Neoadjuvant Immune-Checkpoint Blockade in Triple-Negative Breast Cancer: Current Evidence and Literature-Based Meta-Analysis of Randomized Trials. Cancers 2020, 12, 2497. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Lin, L.T.; Hou, M.F.; Chu, P.Y. PDL1/PD1 blockade in breast cancer: The immunotherapy era (Review). Oncol. Rep. 2021, 45, 5–12. [Google Scholar] [CrossRef]
- Botti, G.; Collina, F.; Scognamiglio, G.; Rao, F.; Peluso, V.; De Cecio, R.; Piezzo, M.; Landi, G.; De Laurentiis, M.; Cantile, M.; et al. Programmed Death Ligand 1 (PD-L1) Tumor Expression Is Associated with a Better Prognosis and Diabetic Disease in Triple Negative Breast Cancer Patients. Int. J. Mol. Sci. 2017, 18, 459. [Google Scholar] [CrossRef]
- Mori, H.; Kubo, M.; Yamaguchi, R.; Nishimura, R.; Osako, T.; Arima, N.; Okumura, Y.; Okido, M.; Yamada, M.; Kai, M.; et al. The combination of PD-L1 expression and decreased tumor-infiltrating lymphocytes is associated with a poor prognosis in triple-negative breast cancer. Oncotarget 2017, 8, 15584–15592. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Adelaide, J.; Chaffanet, M.; Ali, H.R.; Viens, P.; Caldas, C.; Birnbaum, D.; Bertucci, F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015, 6, 5449–5464. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.I.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2018, 25, 34–42. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Management | Risk of Cancer |
|---|---|---|
| Category 0: Need additional evaluation | Re-evaluation | |
| Category 1: Negative finding | Routine screening | Essentially 0% |
| Category 2: Benign lesion | Routine screening | Essentially 0% |
| Category 3: Probably benign lesion | Short-interval follow up | 0–2% |
| Category 4A: Low suspicion of malignancy | Tissue diagnosis | 2–10% |
| Category 4B: Moderate suspicion of malignancy | Tissue diagnosis | 10–50% |
| Category 4C: High suspicion of malignancy | Tissue diagnosis | 50–95% |
| Category 5: Highly suggestive of malignancy | Tissue diagnosis | ≥95% |
| Category 6: Proven malignancy | Surgical excision | - |
| When T | When N | When M | Clinical Stage |
|---|---|---|---|
| Tis | N0 | M0 | 0 |
| T1 | N0 | M0 | IA |
| T0 | N1mi | M0 | IB |
| T1 | N1mi | M0 | IB |
| T0 | N1 | M0 | IIA |
| T1 | N1 | M0 | IIA |
| T2 | N0 | M0 | IIA |
| T2 | N1 | M0 | IIB |
| T3 | N0 | M0 | IIB |
| T0 | N2 | M0 | IIIA |
| T1 | N2 | M0 | IIIA |
| T2 | N2 | M0 | IIIA |
| T3 | N1 | M0 | IIIA |
| T3 | N2 | M0 | IIIA |
| T4 | N0 | M0 | IIIB |
| T4 | N1 | M0 | IIIB |
| T4 | N2 | M0 | IIIB |
| Any T | N3 | M0 | IIIC |
| Any T | Any N | M1 | IV |
| Signaling Pathway | Target | Target Drugs | Ref. |
|---|---|---|---|
| DNA repair pathway | PARP | Olaparib, Veliparib, Rucaparib, Niraparib, Talazoparib | [93] |
| EGF signaling | EGFR | Cetuximab, Panitumumab, Erlotinib, Gefitinib | [94,95] |
| Angiogenesis pathway | VEGF | Bevacizumab, Ramucirumab, Sunitinib, Sorafenib | [80,95,96] |
| PI3K/AKT/mTOR signaling | PDGF, PI3K, mTOR | Sunitinib, Sorafenib, BEZ235, Everolimus, Bez235 | [96,97] |
| MAPK signaling | ERK, mTOR | Bez235, MEK162 | [98] |
| JAK/STAT signaling | JAK2 | Ruxolitinib | [99] |
| Cell cycle pathway | CHK1 | UNC01, AZD7762, PF477736, SCH900776, LY2606368 | [92] |
| Src tyrosine kinase signaling | Src | Dasatinib | [100,101] |
| Human epidermal growth factor receptor 2 | HER2 | Lapatinib, Neratinib, Tucatinib, Poziotinib, Pyrotinib, Trastuzumab, Pertuzumab | [102,103,104,105,106] |
| Fibroblast growth factor receptor signaling | FGFR | Gefinitib, Afanitib, Erlotinib | [107,108,109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-J.; Tzeng, Y.-D.T.; Chiu, Y.-H.; Lin, H.-Y.; Hou, M.-F.; Chu, P.-Y. Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer. Cancers 2021, 13, 2978. https://doi.org/10.3390/cancers13122978
Li C-J, Tzeng Y-DT, Chiu Y-H, Lin H-Y, Hou M-F, Chu P-Y. Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer. Cancers. 2021; 13(12):2978. https://doi.org/10.3390/cancers13122978
Chicago/Turabian StyleLi, Chia-Jung, Yen-Dun Tony Tzeng, Yi-Han Chiu, Hung-Yu Lin, Ming-Feng Hou, and Pei-Yi Chu. 2021. "Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer" Cancers 13, no. 12: 2978. https://doi.org/10.3390/cancers13122978
APA StyleLi, C. -J., Tzeng, Y. -D. T., Chiu, Y. -H., Lin, H. -Y., Hou, M. -F., & Chu, P. -Y. (2021). Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer. Cancers, 13(12), 2978. https://doi.org/10.3390/cancers13122978