
MOUSSE: Multi-Omics Using Subject-Specific SignaturEs

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
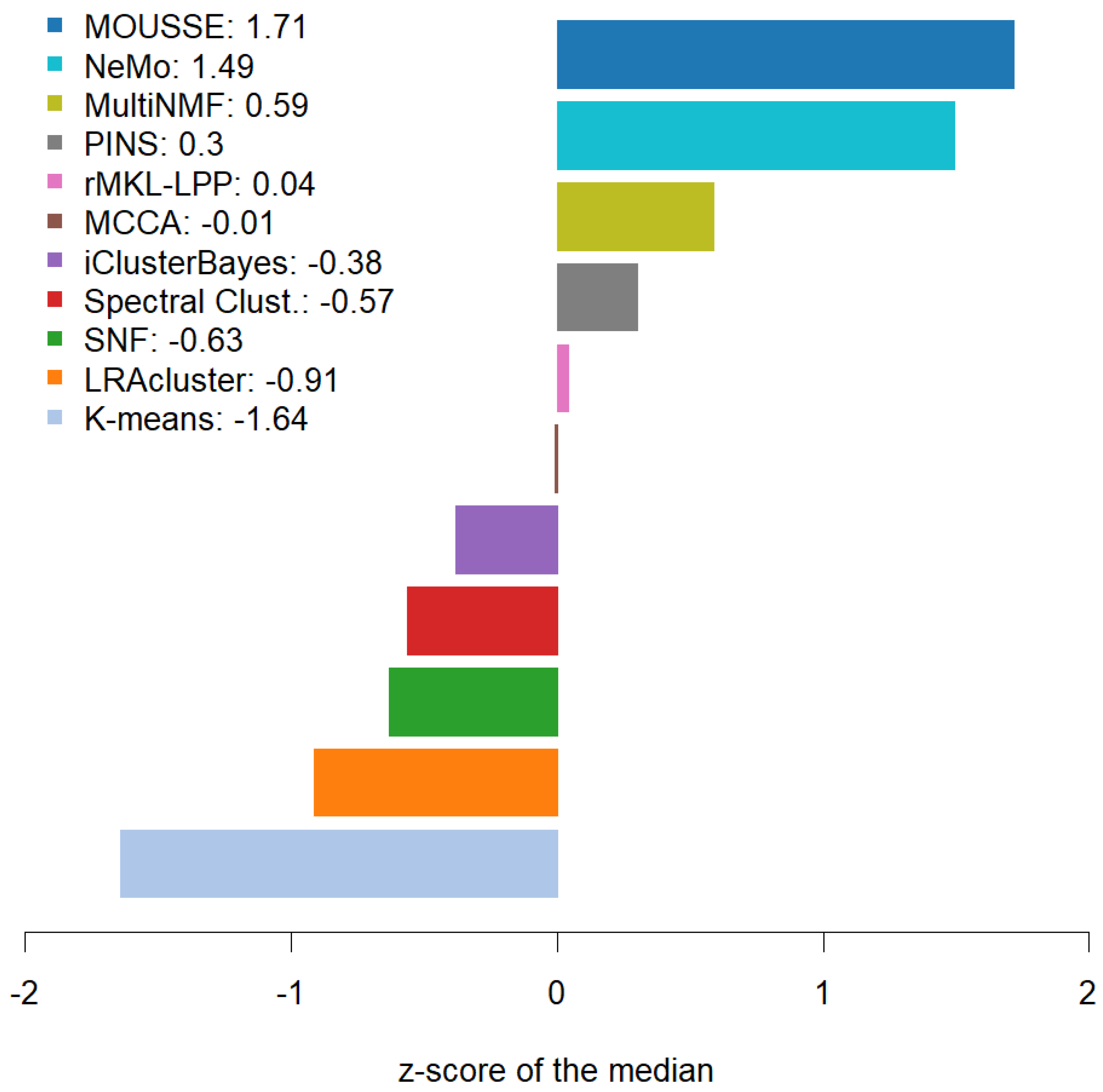
2.1. Benchmark vs. State-of-the-Art Software
2.2. Biological Validation
3. Discussion
3.1. Pipeline Novelties
3.2. Benchmark Results
4. Materials and Methods
4.1. Data Availability
- AML (acute myeloid leukemia);
- BIC (breast invasive carcinoma);
- COAD (colon adenocarcinoma);
- GBM (glioblastoma multiform);
- KIRC (kidney renal clear cell carcinoma);
- LIHC (liver hepatocellular carcinoma);
- LUSC (lung squamous cell carcinoma);
- SKCM (skin cutaneous melanoma);
- SARC (sarcoma);
- OV (ovarian serous cystadenocarcinoma).
4.2. Pipeline Methodology
4.2.1. Preprocessing
4.2.2. Subject-Specific Signature Extraction
4.2.3. Omics-Specific Similarity Networks
4.2.4. Signature Length Optimization
4.2.5. Network Integration and Clustering
4.3. Survival Analysis and Benchmark Comparison
4.4. Biological Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, M.; Tagkopoulos, I. Data integration and predictive modeling methods for multi-omics datasets. Mol. Omics 2017, 14, 8–25. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program-National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 3 December 2020).
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Wspolczesna Onkol. 2015, 1A, A68–A77. [Google Scholar] [CrossRef]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.C.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Wani, N.; Raza, K. Integrative approaches to reconstruct regulatory networks from multi-omics data: A review of state-of-the-art methods. Comput. Biol. Chem. 2019, 83, 107120. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. High Throughput 2019, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Lock, E.F.; Hoadley, K.A.; Marron, J.S.; Nobel, A.B. Joint and individual variation explained (JIVE) for integrated analysis of multiple data types. Ann. Appl. Stat. 2013, 7, 523–542. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef]
- Tini, G.; Marchetti, L.; Priami, C.; Scott-Boyer, M.-P. Multi-omics integration—a comparison of unsupervised clustering methodologies. Briefings Bioinform. 2017, 20, 1269–1279. [Google Scholar] [CrossRef]
- Lauria, M.; Moyseos, P.; Priami, C. SCUDO: A tool for signature-based clustering of expression profiles. Nucleic Acids Res. 2015, 43, W188–W192. [Google Scholar] [CrossRef] [Green Version]
- Hoeng, J.; Stolovitzky, G.; Peitsch, M.C. sbv IMPROVER Diagnostic Signature Challenge. Syst. Biomed. 2013, 1, 193–195. [Google Scholar] [CrossRef]
- Marchetti, L.; Lauria, M.; Caberlotto, L.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E.; Carboni, L. Gene expression signature of antidepressant treatment response/non-response in Flinders Sensitive Line rats subjected to maternal separation. Eur. Neuropsychopharmacol. 2020, 31, 69–85. [Google Scholar] [CrossRef]
- Carboni, L.; Marchetti, L.; Lauria, M.; Gass, P.; Vollmayr, B.; Redfern, A.; Jones, L.; Razzoli, M.; Malki, K.; Begni, V.; et al. Cross-species evidence from human and rat brain transcriptome for growth factor signaling pathway dysregulation in major depression. Neuropsychopharmacology 2018, 43, 2134–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolo, S.; Marchetti, L.; Lauria, M.; Misselbeck, K.; Scott-Boyer, M.-P.; Caberlotto, L.; Priami, C. Combined use of protein biomarkers and network analysis unveils deregulated regulatory circuits in Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0194225. [Google Scholar] [CrossRef]
- Marchetti, L.; Siena, E.; Lauria, M.; Maffione, D.; Pacchiani, N.; Priami, C.; Medini, D. Exploring the Limitations of Peripheral Blood Transcriptional Biomarkers in Predicting Influenza Vaccine Responsiveness. Complexity 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Matone, A.; Derlindati, E.; Marchetti, L.; Spigoni, V.; Cas, A.D.; Montanini, B.; Ardigo, D.; Zavaroni, I.; Priami, C.; Bonadonna, R.C. Identification of an early transcriptomic signature of insulin resistance and related diseases in lymphomonocytes of healthy subjects. PLoS ONE 2017, 12, e0182559. [Google Scholar] [CrossRef] [Green Version]
- Caberlotto, L.; Marchetti, L.; Lauria, M.; Scotti, M.; Parolo, S. Integration of transcriptomic and genomic data suggests candidate mechanisms for APOE4-mediated pathogenic action in Alzheimer’s disease. Sci. Rep. 2016, 6, 32583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, S.; Lauria, M.; Scott-Boyer, M.-P.; Marchetti, L.; Priami, C.; Caberlotto, L. Systems biology approaches to study the molecular effects of caloric restriction and polyphenols on aging processes. Genes Nutr. 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kullback, S.; Leibler, R.A. “On Information and Sufficiency,” The Annals of Mathematical Statistics. Ann. Math. Statist. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Webber, W.; Moffat, A.; Zobel, J. A similarity measure for indefinite rankings. ACM Trans. Inf. Syst. 2010, 28, 1–38. [Google Scholar] [CrossRef]
- Bickel, S.; Scheffer, T. Multi-View Clustering. In Proceedings of the Fourth IEEE International Conference on Data Mining (ICDM ’04), IEEE Computer Society, Washington, DC, USA, 1 November 2004. [Google Scholar]
- Demmel, J. CS267: Notes for Lecture 23, April 9, 1999 Graph Partitioning, Part 2. 1999. Available online: http://people.eecs.berkeley.edu/~demmel/cs267/lecture20/lecture20.html (accessed on 2 July 2021).
- De Sa, V.R. Spectral Clustering with Two Views. In Proceedings of the Workshop on Learning with Multiple Views, 22 nd ICML, Bonn, Germany, 7–11 August 2005. [Google Scholar]
- Wu, D.; Wang, D.; Zhang, M.Q.; Gu, J. Fast dimension reduction and integrative clustering of multi-omics data using low-rank approximation: Application to cancer molecular classification. BMC Genomics 2015, 16, 1022. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Tagett, R.; Diaz, D.; Draghici, S. A novel approach for data integration and disease subtyping. Genome Res. 2017, 27, 2025–2039. [Google Scholar] [CrossRef] [PubMed]
- Speicher, N.K.; Pfeifer, N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics 2015, 31, i268–i275. [Google Scholar] [CrossRef] [Green Version]
- Witten, D.M.; Tibshirani, R.J. Extensions of Sparse Canonical Correlation Analysis with Applications to Genomic Data. Stat. Appl. Genet. Mol. Biol. 2009, 8, 28. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Gao, J.; Han, J. Multi-View Clustering via Joint Nonnegative Matrix Factorization. In Proceedings of the 2013 SIAM International Conference on Data Mining, Society for Industrial and Applied Mathematics, Austin, TX, USA, 2–4 May 2013; pp. 252–260. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Shen, R.; Guo, C.; Vannucci, M.; Chan, K.S.; Hilsenbeck, S.G. A fully Bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics 2018, 19, 71–86. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. NEMO: Cancer subtyping by integration of partial multi-omic data. Bioinformatics 2019, 35, 3348–3356. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Ding, S.; Xu, X.; Nie, R. The latest research progress on spectral clustering. Neural Comput. Appl. 2014, 24, 1477–1486. [Google Scholar] [CrossRef]
- Huang, G.T.; Cunningham, K.I.; Benos, P.V.; Chennubhotla, C.S. Spectral clustering strategies for heterogeneous disease expression data. Pac. Symp. Biocomput. 2013, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Nouri, N.; Kleinstein, S.H. A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics 2018, 34, i341–i349. [Google Scholar] [CrossRef]
- Tarca, A.L.; Lauria, M.; Unger, M.; Bilal, E.; Boue, S.; Dey, K.K.; Hoeng, J.; Koeppl, H.; Martin, F.; Meyer, P.; et al. Strengths and limitations of microarray-based phenotype prediction: Lessons learned from the IMPROVER Diagnostic Signature Challenge. Bioinformatics 2013, 29, 2892–2899. [Google Scholar] [CrossRef]
- Marbach, D.; Costello, J.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manvati, M.K.S.; Khan, J.; Verma, N.; Dhar, P.K. Association of miR-760 with cancer: An overview. Gene 2020, 747, 144648. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhang, R.; Chen, X.; Mu, Y.; Ai, J.; Shi, C.; Liu, Y.; Shi, C.; Sun, L.; Rainov, N.G.; et al. MiR-106a inhibits glioma cell growth by targeting E2F1 independent of p53 status. J. Mol. Med. 2011, 89, 1037–1050. [Google Scholar] [CrossRef]
- Tian, C.; Wu, H.; Li, C.; Tian, X.; Sun, Y.; Liu, E.; Liao, X.; Song, W. Downreguation of FoxM1 by miR-214 inhibits proliferation and migration in hepatocellular carcinoma. Gene Ther. 2018, 25, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-J.; Liu, F.-Y.; Zhang, A.-H.; Liang, H.-F.; Wang, Y.; Ma, R.; Jiang, Y.-H.; Sun, N.-F. MicroRNA-199b-5p attenuates TGF-β1-induced epithelial–mesenchymal transition in hepatocellular carcinoma. Br. J. Cancer 2017, 117, 233–244. [Google Scholar] [CrossRef]
- Rui, T.; Xu, S.; Feng, S.; Zhang, X.; Huang, H.; Ling, Q. The mir-767-105 cluster: A crucial factor related to the poor prognosis of hepatocellular carcinoma. Biomark. Res. 2020, 8, 7. [Google Scholar] [CrossRef]
- Zhang, R.; Pang, B.; Xin, T.; Guo, H.; Xing, Y.; Xu, S.; Feng, B.; Liu, B.; Pang, Q. Plasma miR-221/222 Family as Novel Descriptive and Prognostic Biomarkers for Glioma. Mol. Neurobiol. 2016, 53, 1452–1460. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, X.; Sun, C.; Guo, H.; Wang, T.; Wei, L.; Zhang, Y.; Zhao, J.; Ma, X. LncRNA LINC00963 Promotes Tumorigenesis and Radioresistance in Breast Cancer by Sponging miR-324-3p and Inducing ACK1 Expression. Mol. Ther. Nucleic Acids 2019, 18, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, J.; Zhang, X.; Fu, B.; Song, Y.; Ma, P.; Gu, K.; Zhou, X.; Zhang, X.; Tian, T.; et al. N6-Methyladenine hinders RNA- and DNA-directed DNA synthesis: Application in human rRNA methylation analysis of clinical specimens. Chem. Sci. 2016, 7, 1440–1446. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Zheng, L.; Sun, D.; Li, X.; Xu, P.; Tian, L. miR-204 functions as a tumor suppressor gene, at least partly by suppressing CYP27A1 in glioblastoma. Oncol. Lett. 2018, 16, 1439–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songsheng, S.; Fajol, A.; Tu, X.; Ren, B.; Shi, S. miR-204 suppresses the development and progression of human glioblastoma by targeting ATF2. Oncotarget 2016, 7, 70058–70065. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gong, X.; Chen, J.; Zhang, J.; Sun, J.; Guo, M. miR-340 inhibits glioblastoma cell proliferation by suppressing CDK6, cyclin-D1 and cyclin-D2. Biochem. Biophys. Res. Commun. 2015, 460, 670–677. [Google Scholar] [CrossRef]
- Hu, S.-H.; Wang, C.-H.; Huang, Z.-J.; Liu, F.; Xu, C.-W.; Li, X.-L.; Chen, G.-Q. miR-760 mediates chemoresistance through inhibition of epithelial mesenchymal transition in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5002–5008. [Google Scholar]
- Miao, Y.; Zheng, W.; Li, N.; Su, Z.; Zhao, L.; Zhou, H.; Jia, L. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 41942. [Google Scholar] [CrossRef]
- Lan, F.; Qing, Q.; Pan, Q.; Huiming, Y.; Yu, H.; Yue, X. Serum exosomal miR-301a as a potential diagnostic and prognostic biomarker for human glioma. Cell. Oncol. 2018, 41, 25–33. [Google Scholar] [CrossRef]
- El-Abd, N.E.; Fawzy, N.A.; El-Sheikh, S.M.; Soliman, M.E. Circulating miRNA-122, miRNA-199a, and miRNA-16 as Biomarkers for Early Detection of Hepatocellular Carcinoma in Egyptian Patients with Chronic Hepatitis C Virus Infection. Mol. Diagn. Ther. 2015, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Liao, Y.; Jin, C.; Zhang, Z.; Zhao, J.; Liu, K.; Huang, H.; Cao, H.; Cheng, Q. Identification of SEC61G as a Novel Prognostic Marker for Predicting Survival and Response to Therapies in Patients with Glioblastoma. Med. Sci. Monit. 2019, 25, 3624–3635. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Liu, Q.; Peng, G.; Su, J.; Qin, C.-Y.; Wang, X.-Y. Identification and validation of a three-gene signature as a candidate prognostic biomarker for lower grade glioma. PeerJ 2020, 8, e8312. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Kawaguchi, A.; Kanayama, T.; Hayano, A.; Yamanaka, R. Correlation between lower balance of Th2 helper T-cells and expression of PD-L1/PD-1 axis genes enables prognostic prediction in patients with glioblastoma. Oncotarget 2018, 9, 19065–19078. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Tang, Z.; Yang, Z.; Zhang, L.; Deng, Q.; Zhang, X.; Yu, Y.; Liu, X.; Zhu, J. EXO1 overexpression is associated with poor prognosis of hepatocellular carcinoma patients. Cell Cycle 2018, 17, 2386–2397. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Rostomily, R.; Wong, S.T.C. Prognostic Gene Discovery in Glioblastoma Patients using Deep Learning. Cancers 2019, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Sun, J.; Chen, X.; Liu, L.; Wu, D. Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of β-catenin in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 316. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, H.; Park, D.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Jeoung, D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010, 285, 25957–25968. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.; Ratzinger, S.; Wales, L.; Bosserhoff, A.; Senner, V.; Grifka, J.; Grässel, S. Inhibition of Collagen XVI Expression Reduces Glioma Cell Invasiveness. Cell. Physiol. Biochem. 2011, 27, 217–226. [Google Scholar] [CrossRef]
- Sahu, S.K.; Fritz, A.; Tiwari, N.; Kovacs, Z.; Pouya, A.; Wüllner, V.; Bora, P.; Schacht, T.; Baumgart, J.; Peron, S.; et al. TOX3 regulates neural progenitor identity. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 833–840. [Google Scholar] [CrossRef]
- Jin, X.; Jin, X.; Jung, J.-E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef]
- Dougherty, J.D.; Fomchenko, E.I.; Akuffo, A.A.; Schmidt, E.; Helmy, K.Y.; Bazzoli, E.; Brennan, C.W.; Holland, E.C.; Milosevic, A. Candidate Pathways for Promoting Differentiation or Quiescence of Oligodendrocyte Progenitor-like Cells in Glioma. Cancer Res. 2012, 72, 4856–4868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laks, D.; Masterman-Smith, M.; Visnyei, K.; Angenieux, B.; Orozco, N.M.; Foran, I.; Yong, W.H.; Vinters, H.V.; Liau, L.; Lazareff, J.A.; et al. Neurosphere Formation Is an Independent Predictor of Clinical Outcome in Malignant Glioma. Stem Cells 2009, 27, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. Abstract 3287: An integrated TCGA pan-cancer clinical data resource to drive high quality survival outcome analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}

| miRNA | Genes | ||||
|---|---|---|---|---|---|
| BIC | GBM | LIHC | BIC | GBM | LIHC |
| let-7c | miR-222 | mir-105-2 | LOC728264 | TOX3 | DSCR4 |
| mir-140 | miR-23a | mir-767 | SLC7A3 | SEC61G | SSX6 |
| mir-1307 | miR-204 | mir-105-1 | HSPD1 | C20orf42 | EXO1 |
| mir-101-2 | miR-34b | mir-139 | IGFN1 | PLA2G2A | NEK2 |
| mir-33b | miR-221 | mir-199a-1 | AURKA | CRTAC1 | RHOXF2B |
| mir-99b | miR-340 | mir-199a-2 | ANGPTL7 | CA10 | DCAF8L1 |
| mir-324 | miR-181a* | mir-10a | TPX2 | GPR17 | PAGE2 |
| mir-760 | miR-17-5p | mir-214 | CCL16 | COL16A1 | RNF17 |
| mir-130b | miR-106a | mir-199b | SGOL1 | MAB21L1 | DDX53 |
| mir-331 | miR-301 | mir-22 | NPY2R | SLC11A1 | MAGEB16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorentino, G.; Visintainer, R.; Domenici, E.; Lauria, M.; Marchetti, L. MOUSSE: Multi-Omics Using Subject-Specific SignaturEs. Cancers 2021, 13, 3423. https://doi.org/10.3390/cancers13143423
Fiorentino G, Visintainer R, Domenici E, Lauria M, Marchetti L. MOUSSE: Multi-Omics Using Subject-Specific SignaturEs. Cancers. 2021; 13(14):3423. https://doi.org/10.3390/cancers13143423
Chicago/Turabian StyleFiorentino, Giuseppe, Roberto Visintainer, Enrico Domenici, Mario Lauria, and Luca Marchetti. 2021. "MOUSSE: Multi-Omics Using Subject-Specific SignaturEs" Cancers 13, no. 14: 3423. https://doi.org/10.3390/cancers13143423
APA StyleFiorentino, G., Visintainer, R., Domenici, E., Lauria, M., & Marchetti, L. (2021). MOUSSE: Multi-Omics Using Subject-Specific SignaturEs. Cancers, 13(14), 3423. https://doi.org/10.3390/cancers13143423