
The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer




Abstract
:Simple Summary
Abstract
1. Introduction
2. Vascular Normalization and Tumor Hypoxia
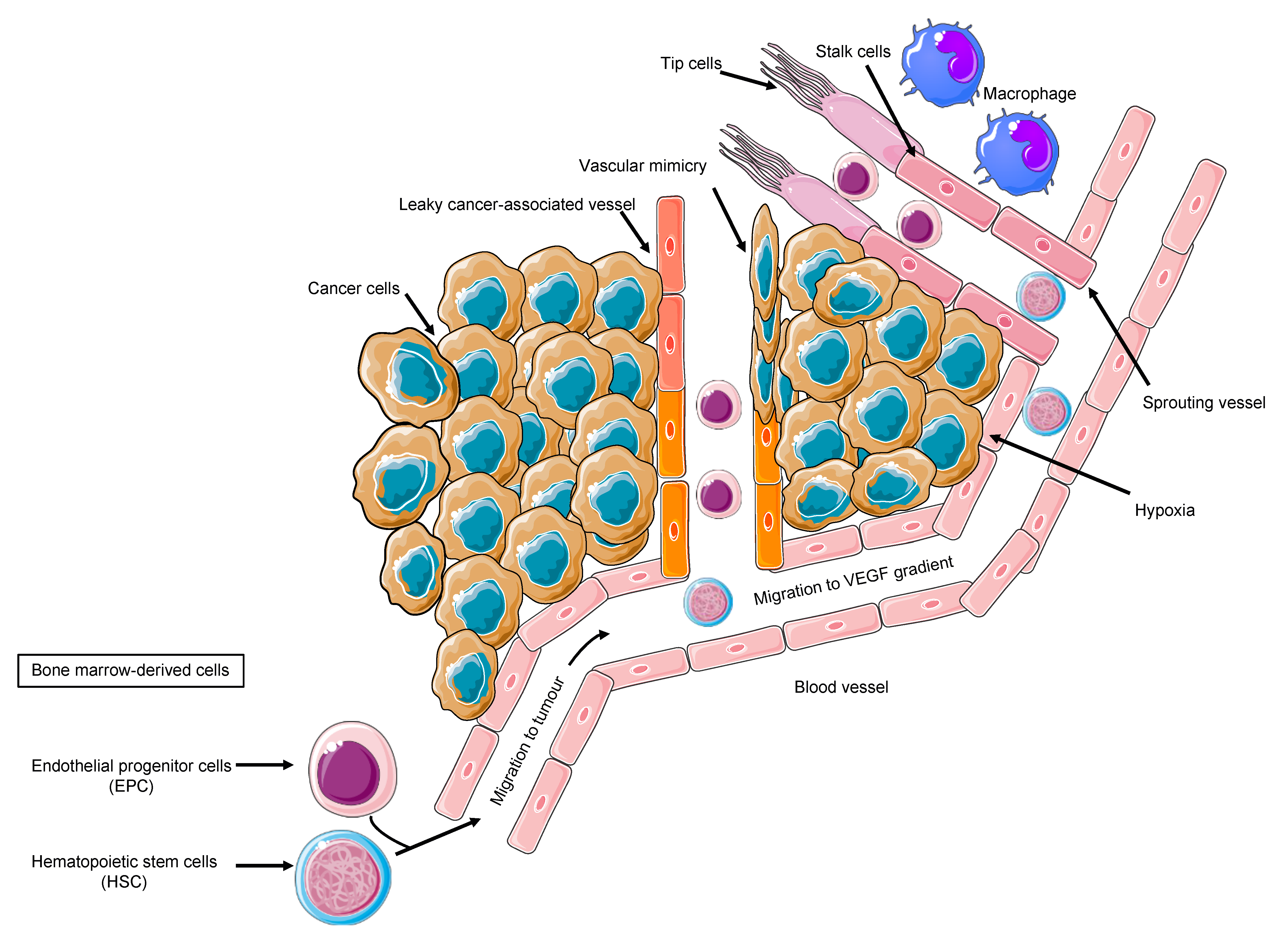
3. Role of Inflammatory Cells, Endothelial Cells, and Tumor Cells
4. Alternative Mechanisms of Tumor Vasculature
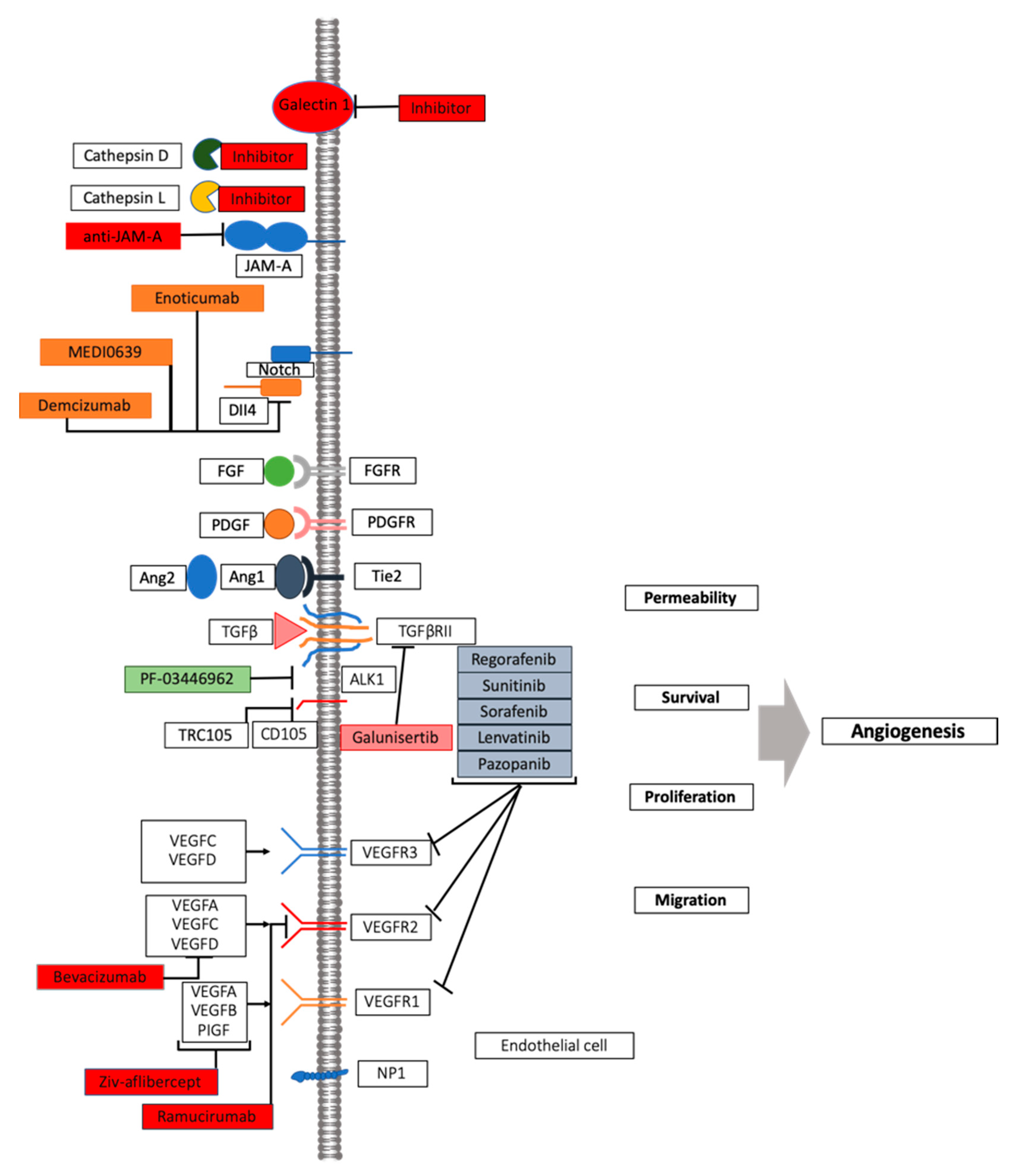
5. Pleiotropic Role of VEGF in Angiogenesis, Inflammation, and Immunity
6. The Biology of Angiogenesis and Its Implication in Overcoming the Challenge of Identifying Biomarkers
7. What Can We Do and What Needs to Be Done?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marmé, D. Tumor Angiogenesis A Key Target for Cancer Therapy; Springer: Singapore, 2020; ISBN 978-3-319-31215-6. [Google Scholar]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.T.; Haap, M.; Kopp, H.-G.; Lipp, H.-P. Tyrosine Kinase Inhibitors—A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Gaya, A.; Tse, V. A Preclinical and Clinical Review of Aflibercept for the Management of Cancer. Cancer Treat. Rev. 2012, 38, 484–493. [Google Scholar] [CrossRef]
- Van der Veldt, A.A.M.; Lubberink, M.; Bahce, I.; Walraven, M.; de Boer, M.P.; Greuter, H.N.J.M.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid Decrease in Delivery of Chemotherapy to Tumors after Anti-VEGF Therapy: Implications for Scheduling of Anti-Angiogenic Drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.R.; Davis, R.; Norberg, S.M.; O’Brien, S.; Sennino, B.; Nakahara, T.; Yao, V.J.; Inai, T.; Brooks, P.; Freimark, B.; et al. Rapid Vascular Regrowth in Tumors after Reversal of VEGF Inhibition. J. Clin. Investig. 2006, 116, 2610–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothey, A.; Sugrue, M.M.; Purdie, D.M.; Dong, W.; Sargent, D.; Hedrick, E.; Kozloff, M. Bevacizumab beyond First Progression Is Associated with Prolonged Overall Survival in Metastatic Colorectal Cancer: Results from a Large Observational Cohort Study (BRiTE). J. Clin. Oncol. 2008, 26, 5326–5334. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Pantano, F.; Iuliani, M.; Santini, D.; Silvestris, N.; Vacca, A. Skeletal Metastases of Unknown Primary: Biological Landscape and Clinical Overview. Cancers 2019, 11, 1270. [Google Scholar] [CrossRef] [Green Version]
- Antonio, G.; Oronzo, B.; Vito, L.; Angela, C.; Antonel-La, A.; Roberto, C.; Giovanni, S.A.; Antonella, L. Immune System and Bone Microenvironment: Rationale for Targeted Cancer Therapies. Oncotarget 2020, 11, 480. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-Angiogenesis and Immunotherapy: Novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Kaelin, W.G. The VHL/HIF Axis in Clear Cell Renal Carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Goldman, C.K.; Kim, J.; Wong, W.L.; King, V.; Brock, T.; Gillespie, G.Y. Epidermal Growth Factor Stimulates Vascular Endothelial Growth Factor Production by Human Malignant Glioma Cells: A Model of Glioblastoma Multiforme Pathophysiology. Mol. Biol. Cell 1993, 4, 121–133. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The Great Escape; the Hallmarks of Resistance to Antiangiogenic Therapy. Pharm. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [Green Version]
- Huijbers, E.J.M.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the Tumor Stroma in Resistance to Anti-Angiogenic Therapy. Drug Resist. Updat. 2016, 25, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting Hypoxia in Cancer Therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finger, E.C.; Giaccia, A.J. Hypoxia, Inflammation, and the Tumor Microenvironment in Metastatic Disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Summa, S.D.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef]
- Sounni, N.E.; Cimino, J.; Blacher, S.; Primac, I.; Truong, A.; Mazzucchelli, G.; Paye, A.; Calligaris, D.; Debois, D.; De Tullio, P.; et al. Blocking Lipid Synthesis Overcomes Tumor Regrowth and Metastasis after Antiangiogenic Therapy Withdrawal. Cell Metab. 2014, 20, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Vandyke, K.; Zeissig, M.N.; Hewett, D.R.; Martin, S.K.; Mrozik, K.M.; Cheong, C.M.; Diamond, P.; To, L.B.; Gronthos, S.; Peet, D.J.; et al. HIF-2α Promotes Dissemination of Plasma Cells in Multiple Myeloma by Regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017, 77, 5452–5463. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Vacca, A.; Ribatti, D. A Comprehensive Biological and Clinical Perspective Can Drive a Patient-Tailored Approach to Multiple Myeloma: Bridging the Gaps between the Plasma Cell and the Neoplastic Niche. J. Oncol. 2020, 2020, 6820241. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic Control of Metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.-H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; van Bruggen, N.; et al. Bv8 Regulates Myeloid-Cell-Dependent Tumour Angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, O.; Biesiekierska, M.; Panthu, B.; Vialichka, V.; Pirola, L.; Balcerczyk, A. The Epigenetic Profile of Tumor Endothelial Cells. Effects of Combined Therapy with Antiangiogenic and Epigenetic Drugs on Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2606. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Hou, L.; Li, J.; Shao, S.; Huang, S.; Meng, D.; Liu, L.; Feng, L.; Xia, P.; Qin, T.; et al. VEGF/NRP-1axis Promotes Progression of Breast Cancer via Enhancement of Epithelial-Mesenchymal Transition and Activation of NF-ΚB and β-Catenin. Cancer Lett. 2016, 373, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Ishida, J.; Kurozumi, K.; Ichikawa, T.; Otani, Y.; Oka, T.; Tomita, Y.; Hattori, Y.; Uneda, A.; Matsumoto, Y.; et al. δ-Catenin Promotes Bevacizumab-Induced Glioma Invasion. Mol. Cancer Ther. 2019, 18, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.-L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene Expression Profile Identifies Tyrosine Kinase C-Met as a Targetable Mediator of Antiangiogenic Therapy Resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Djonov, V. Intussusceptive Microvascular Growth in Tumors. Cancer Lett. 2012, 316, 126–131. [Google Scholar] [CrossRef]
- Pezzella, F.; Ribatti, D. Vascular Co-Option and Vasculogenic Mimicry Mediate Resistance to Antiangiogenic Strategies. Cancer Rep. 2020, e1318. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Reynolds, A.R.; Kuczynski, E.A.; Gatter, K.; Vermeulen, P.B.; Kerbel, R.S.; Harris, A.L.; Pezzella, F. Non-Angiogenic Tumours and Their Influence on Cancer Biology. Nat. Rev. Cancer 2018, 18, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.B.; Hendrix, M.J.C. Molecular Pathways: Vasculogenic Mimicry in Tumor Cells: Diagnostic and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, Q.; Li, X.-Y.; Yang, Q.-Y.; Xu, W.-W.; Liu, G.-L. Short-Term Anti-Vascular Endothelial Growth Factor Treatment Elicits Vasculogenic Mimicry Formation of Tumors to Accelerate Metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 16. [Google Scholar] [CrossRef] [Green Version]
- Finley, S.D.; Chu, L.-H.; Popel, A.S. Computational Systems Biology Approaches to Anti-Angiogenic Cancer Therapeutics. Drug Discov. Today 2015, 20, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.; Danesi, R.; Agen, C.; Di Paolo, A.; Basolo, F.; Del Bianchi, S.; Del Tacca, M. MCF-10A Cells Infected with the Int-2 Oncogene Induce Angiogenesis in the Chick Chorioallantoic Membrane and in the Rat Mesentery. Cancer Res. 1994, 54, 9–11. [Google Scholar]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous Embryonic Lethality Induced by Targeted Inactivation of the VEGF Gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Roy, H.; Bhardwaj, S.; Ylä-Herttuala, S. Biology of Vascular Endothelial Growth Factors. FEBS Lett. 2006, 580, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Verdegem, D.; Moens, S.; Stapor, P.; Carmeliet, P. Endothelial Cell Metabolism: Parallels and Divergences with Cancer Cell Metabolism. Cancer Metab. 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Krebs, M.; Solimando, A.G.; Kalogirou, C.; Marquardt, A.; Frank, T.; Sokolakis, I.; Hatzichristodoulou, G.; Kneitz, S.; Bargou, R.; Kübler, H.; et al. MiR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. J. Clin. Med. 2020, 9, 670. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Chen, X. Multimodality Molecular Imaging of Tumor Angiogenesis. J. Nucl. Med. 2008, 49, 113S–128S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia Promotes Tumor Growth in Linking Angiogenesis to Immune Escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Yamashita-Kashima, Y.; Fujimoto-Ouchi, K.; Yorozu, K.; Kurasawa, M.; Yanagisawa, M.; Yasuno, H.; Mori, K. Biomarkers for Antitumor Activity of Bevacizumab in Gastric Cancer Models. BMC Cancer 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-T.; Yuan, X.-W.; Zhu, H.-T.; Deng, Z.-M.; Yu, D.-C.; Zhou, X.; Ding, Y.-T. Endothelial Precursor Cells Promote Angiogenesis in Hepatocellular Carcinoma. World J. Gastroenterol. 2012, 18, 4925–4933. [Google Scholar] [CrossRef] [PubMed]
- Backer, M.V.; Backer, J.M. Imaging Key Biomarkers of Tumor Angiogenesis. Theranostics 2012, 2, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laking, G.R.; West, C.; Buckley, D.L.; Matthews, J.; Price, P.M. Imaging Vascular Physiology to Monitor Cancer Treatment. Crit. Rev. Oncol. Hematol. 2006, 58, 95–113. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Linehan, W.M.; Vasselli, J.; Srinivasan, R.; Walther, M.M.; Merino, M.; Choyke, P.; Vocke, C.; Schmidt, L.; Isaacs, J.S.; Glenn, G.; et al. Genetic Basis of Cancer of the Kidney: Disease-Specific Approaches to Therapy. Clin. Cancer Res. 2004, 10, 6282S–6289S. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Z.; Zhang, H.; Jin, H.; Sun, L.; Dong, H.; Xu, M.; Zhao, P.; Zhang, B.; Wang, J.; et al. HIF-1α and HIF-2α Correlate with Migration and Invasion in Gastric Cancer. Cancer Biol. Ther. 2010, 10, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Tanigawa, N.; Amaya, H.; Matsumura, M.; Shimomatsuya, T. Correlation between Expression of Vascular Endothelial Growth Factor and Tumor Vascularity, and Patient Outcome in Human Gastric Carcinoma. J. Clin. Oncol. 1997, 15, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Youssoufian, H.; Hicklin, D.J.; Rowinsky, E.K. Review: Monoclonal Antibodies to the Vascular Endothelial Growth Factor Receptor-2 in Cancer Therapy. Clin. Cancer Res. 2007, 13, 5544s–5548s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Chung, Y.S.; Ogawa, Y.; Takatsuka, S.; Kang, S.M.; Ogawa, M.; Sawada, T.; Sowa, M. Prognostic Value of Vascular Endothelial Growth Factor Expression in Gastric Carcinoma. Cancer 1996, 77, 858–863. [Google Scholar] [CrossRef]
- Suzuki, S.; Dobashi, Y.; Hatakeyama, Y.; Tajiri, R.; Fujimura, T.; Heldin, C.H.; Ooi, A. Clinicopathological Significance of Platelet-Derived Growth Factor (PDGF)-B and Vascular Endothelial Growth Factor-A Expression, PDGF Receptor-β Phosphorylation, and Microvessel Density in Gastric Cancer. BMC Cancer 2010, 10, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarejo-Campos, P.; Padilla-Valverde, D.; Martin, R.M.; Menéndez-Sánchez, P.; Cubo-Cintas, T.; Bondia-Navarro, J.A.; Fernández, J.M. Serum VEGF and VEGF-C Values before Surgery and after Postoperative Treatment in Gastric Cancer. Clin. Transl. Oncol. 2013, 15, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Fang, J.; Yang, C. Overexpression of Both VEGF-A and VEGF-C in Gastric Cancer Correlates with Prognosis, and Silencing of Both Is Effective to Inhibit Cancer Growth. Int. J. Clin. Exp. Pathol. 2013, 6, 586–597. [Google Scholar]
- Yanagisawa, M.; Yorozu, K.; Kurasawa, M.; Nakano, K.; Furugaki, K.; Yamashita, Y.; Mori, K.; Fujimoto-Ouchi, K. Bevacizumab Improves the Delivery and Efficacy of Paclitaxel. Anticancer Drugs 2010, 21, 687–694. [Google Scholar] [CrossRef]
- Kamiyama, H.; Takano, S.; Tsuboi, K.; Matsumura, A. Anti-Angiogenic Effects of SN38 (Active Metabolite of Irinotecan): Inhibition of Hypoxia-Inducible Factor 1 Alpha (HIF-1alpha)/Vascular Endothelial Growth Factor (VEGF) Expression of Glioma and Growth of Endothelial Cells. J. Cancer Res. Clin. Oncol. 2005, 131, 205–213. [Google Scholar] [CrossRef]
- Giuliano, S.; Pagès, G. Mechanisms of Resistance to Anti-Angiogenesis Therapies. Biochimie 2013, 95, 1110–1119. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid Biopsy and Tumor Heterogeneity in Metastatic Solid Tumors: The Potentiality of Blood Samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Méndez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; García-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-Dependent Lectin-Receptor Interactions Preserve Angiogenesis in Anti-VEGF Refractory Tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Hanahan, D. Modes of Resistance to Anti-Angiogenic Therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, H.; Falcón, B.L.; Kuroda, T.; Baluk, P.; Coxon, A.; Yu, D.; Bready, J.V.; Oliner, J.D.; McDonald, D.M. Complementary Actions of Inhibitors of Angiopoietin-2 and VEGF on Tumor Angiogenesis and Growth. Cancer Res. 2010, 70, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of MTOR Complex 2 Restrains Tumor Angiogenesis in Multiple Myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Giannico, D.; Leone, P.; Solimando, A.G.; Maiorano, E.; Caporusso, C.; Duda, L.; Tamma, R.; Mallamaci, R.; Susca, N.; et al. HB-EGF-EGFR Signaling in Bone Marrow Endothelial Cells Mediates Angiogenesis Associated with Multiple Myeloma. Cancers 2020, 12, 173. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting Angiogenesis in Multiple Myeloma by the VEGF and HGF Blocking DARPin® Protein MP0250: A Preclinical Study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Buonavoglia, A.; Fasano, R.; Solimando, A.G.; De Re, V.; Cicco, S.; Vacca, A.; Racanelli, V. Insights into the Regulation of Tumor Angiogenesis by Micro-RNAs. J. Clin. Med. 2019, 8, 2030. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016, 37, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Jridi, I.; Catacchio, I.; Majdoub, H.; Shahbazzadeh, D.; El Ayeb, M.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Vacca, A.; Borchani, L. The Small Subunit of Hemilipin2, a New Heterodimeric Phospholipase A2 from Hemiscorpius Lepturus Scorpion Venom, Mediates the Antiangiogenic Effect of the Whole Protein. Toxicon 2017, 126, 38–46. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic Therapy in Oncology: Current Status and Future Directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Tartour, E.; Pere, H.; Maillere, B.; Terme, M.; Merillon, N.; Taieb, J.; Sandoval, F.; Quintin-Colonna, F.; Lacerda, K.; Karadimou, A.; et al. Angiogenesis and Immunity: A Bidirectional Link Potentially Relevant for the Monitoring of Antiangiogenic Therapy and the Development of Novel Therapeutic Combination with Immunotherapy. Cancer Metastasis Rev. 2011, 30, 83–95. [Google Scholar] [CrossRef]
- Korc, M. Pathways for Aberrant Angiogenesis in Pancreatic Cancer. Mol. Cancer 2003, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11, 330. [Google Scholar] [CrossRef] [Green Version]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Paul, S.; Mancuso, P.; Monestiroli, S.; Gobbi, A.; Shaked, Y.; Kerbel, R.S. Maximum Tolerable Dose and Low-Dose Metronomic Chemotherapy Have Opposite Effects on the Mobilization and Viability of Circulating Endothelial Progenitor Cells. Cancer Res. 2003, 63, 4342–4346. [Google Scholar] [PubMed]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-Beta in Recruitment of Vascular Smooth Muscle Cells and Pericytes during Embryonic Blood Vessel Formation in the Mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Betsholtz, C. Endothelial-Pericyte Interactions in Angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef]
- Chintalgattu, V.; Rees, M.L.; Culver, J.C.; Goel, A.; Jiffar, T.; Zhang, J.; Dunner, K.; Pati, S.; Bankson, J.A.; Pasqualini, R.; et al. Coronary Microvascular Pericytes Are the Cellular Target of Sunitinib Malate-Induced Cardiotoxicity. Sci. Transl. Med. 2013, 5, 187ra69. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.L.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or Fueling Metastasis with VEGF Inhibitors: Antiangiogenesis Revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, R.M.; Torcuator, R.; Jain, R.; Anderson, J.; Doyle, T.; Ellika, S.; Schultz, L.; Mikkelsen, T. Efficacy, Safety and Patterns of Response and Recurrence in Patients with Recurrent High-Grade Gliomas Treated with Bevacizumab plus Irinotecan. J. Neurooncol. 2009, 91, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular Vesicles in Angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to Sunitinib in Renal Clear Cell Carcinoma Results from Sequestration in Lysosomes and Inhibition of the Autophagic Flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Marinoni, I.; Perren, A. Autophagy Inhibition Improves Sunitinib Efficacy in Pancreatic Neuroendocrine Tumors via a Lysosome-Dependent Mechanism. Mol. Cancer Ther. 2017, 16, 2502–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Duda, D.G.; Willett, C.G.; Sahani, D.V.; Zhu, A.X.; Loeffler, J.S.; Batchelor, T.T.; Sorensen, A.G. Biomarkers of Response and Resistance to Antiangiogenic Therapy. Nat. Rev. Clin. Oncol. 2009, 6, 327–338. [Google Scholar] [CrossRef]
- Lambrechts, D.; Lenz, H.-J.; de Haas, S.; Carmeliet, P.; Scherer, S.J. Markers of Response for the Antiangiogenic Agent Bevacizumab. J. Clin. Oncol. 2013, 31, 1219–1230. [Google Scholar] [CrossRef]
- Arevalo, O.D.; Soto, C.; Rabiei, P.; Kamali, A.; Ballester, L.Y.; Esquenazi, Y.; Zhu, J.-J.; Riascos, R.F. Assessment of Glioblastoma Response in the Era of Bevacizumab: Longstanding and Emergent Challenges in the Imaging Evaluation of Pseudoresponse. Front. Neurol. 2019, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Jubb, A.M.; Chen, D.; Li, N.F.; Meng, Y.G.; Bernaards, C.; Elliott, R.; Scherer, S.J.; Chen, D.S. Predictive Impact of Circulating Vascular Endothelial Growth Factor in Four Phase III Trials Evaluating Bevacizumab. Clin. Cancer Res. 2013, 19, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, D.W.; de Haas, S.L.; Dirix, L.Y.; Romieu, G.; Chan, A.; Pivot, X.; Tomczak, P.; Provencher, L.; Cortés, J.; Delmar, P.R.; et al. Biomarker Results from the AVADO Phase 3 Trial of First-Line Bevacizumab plus Docetaxel for HER2-Negative Metastatic Breast Cancer. Br. J. Cancer 2013, 108, 1052–1060. [Google Scholar] [CrossRef]
- Gianni, L.; Romieu, G.H.; Lichinitser, M.; Serrano, S.V.; Mansutti, M.; Pivot, X.; Mariani, P.; Andre, F.; Chan, A.; Lipatov, O.; et al. AVEREL: A Randomized Phase III Trial Evaluating Bevacizumab in Combination with Docetaxel and Trastuzumab as First-Line Therapy for HER2-Positive Locally Recurrent/Metastatic Breast Cancer. J. Clin. Oncol. 2013, 31, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Cameron, D.; Bondarenko, I.; Manzyuk, L.; Alcedo, J.C.; Lopez, R.I.; Im, S.-A.; Canon, J.-L.; Shparyk, Y.; Yardley, D.A.; et al. Bevacizumab plus Paclitaxel versus Placebo plus Paclitaxel as First-Line Therapy for HER2-Negative Metastatic Breast Cancer (MERiDiAN): A Double-Blind Placebo-Controlled Randomised Phase III Trial with Prospective Biomarker Evaluation. Eur. J. Cancer 2017, 70, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Takahashi, M.; Nakagami, K.; Okumura, Y.; Nakayama, T.; Sato, N.; Kanatani, K.; Tajima, K.; Kashiwaba, M. First-Line Bevacizumab plus Paclitaxel in Japanese Patients with HER2-Negative Metastatic Breast Cancer: Subgroup Results from the Randomized Phase III MERiDiAN Trial. Jpn. J. Clin. Oncol. 2017, 47, 385–392. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination with Bevacizumab versus Sunitinib in Renal Cell Carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the Vicious Cycle within the Multiple Myeloma Ecosystem: Blocking JAM-A on Bone Marrow Endothelial Cells Restores the Angiogenic Homeostasis and Suppresses Tumor Progression. Haematologica 2021, 106, 1943–1956. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Annese, T.; Tamma, R.; Ingravallo, G.; Maiorano, E.; Vacca, A.; Specchia, G.; Ribatti, D. New Insights into Diffuse Large B-Cell Lymphoma Pathobiology. Cancers 2020, 12, 1869. [Google Scholar] [CrossRef]
- Rapisarda, A.; Melillo, G. Role of the Hypoxic Tumor Microenvironment in the Resistance to Anti-Angiogenic Therapies. Drug Resist. Updat. 2009, 12, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrechts, D.; Thienpont, B.; Thuillier, V.; Sagaert, X.; Moisse, M.; Peuteman, G.; Pericay, C.; Folprecht, G.; Zalcberg, J.; Zilocchi, C.; et al. Evaluation of Efficacy and Safety Markers in a Phase II Study of Metastatic Colorectal Cancer Treated with Aflibercept in the First-Line Setting. Br. J. Cancer 2015, 113, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Claes, B.; Delmar, P.; Reumers, J.; Mazzone, M.; Yesilyurt, B.T.; Devlieger, R.; Verslype, C.; Tejpar, S.; Wildiers, H.; et al. VEGF Pathway Genetic Variants as Biomarkers of Treatment Outcome with Bevacizumab: An Analysis of Data from the AViTA and AVOREN Randomised Trials. Lancet Oncol. 2012, 13, 724–733. [Google Scholar] [CrossRef]
- Beuselinck, B.; Karadimou, A.; Lambrechts, D.; Claes, B.; Wolter, P.; Couchy, G.; Berkers, J.; Paridaens, R.; Schöffski, P.; Méjean, A.; et al. Single-Nucleotide Polymorphisms Associated with Outcome in Metastatic Renal Cell Carcinoma Treated with Sunitinib. Br. J. Cancer 2013, 108, 887–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuselinck, B.; Karadimou, A.; Lambrechts, D.; Claes, B.; Wolter, P.; Couchy, G.; Berkers, J.; van Poppel, H.; Paridaens, R.; Schöffski, P.; et al. VEGFR1 Single Nucleotide Polymorphisms Associated with Outcome in Patients with Metastatic Renal Cell Carcinoma Treated with Sunitinib—A Multicentric Retrospective Analysis. Acta Oncol. 2014, 53, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Willett, C.G.; Ancukiewicz, M.; di Tomaso, E.; Shah, M.; Czito, B.G.; Bentley, R.; Poleski, M.; Lauwers, G.Y.; Carroll, M.; et al. Plasma Soluble VEGFR-1 Is a Potential Dual Biomarker of Response and Toxicity for Bevacizumab with Chemoradiation in Locally Advanced Rectal Cancer. Oncologist 2010, 15, 577–583. [Google Scholar] [CrossRef]
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, Safety, and Biomarkers of Neoadjuvant Bevacizumab, Radiation Therapy, and Fluorouracil in Rectal Cancer: A Multidisciplinary Phase II Study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef] [Green Version]
- Heist, R.S.; Duda, D.G.; Sahani, D.V.; Ancukiewicz, M.; Fidias, P.; Sequist, L.V.; Temel, J.S.; Shaw, A.T.; Pennell, N.A.; Neal, J.W.; et al. Improved Tumor Vascularization after Anti-VEGF Therapy with Carboplatin and Nab-Paclitaxel Associates with Survival in Lung Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi, Y.; Iinuma, H.; Horiuchi, A.; Shimada, R.; Watanabe, T. Association of Plasma VEGF-A, Soluble VEGFR-1 and VEGFR-2 Levels and Clinical Response and Survival in Advanced Colorectal Cancer Patients Receiving Bevacizumab with Modified FOLFOX6. Oncol. Lett. 2010, 1, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of Vascular Density and Normalization in Response to Neoadjuvant Bevacizumab and Chemotherapy in Breast Cancer Patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Ancukiewicz, M.; Supko, J.G.; Sahani, D.V.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Abrams, T.A.; McCleary, N.J.; Bhargava, P.; Muzikansky, A.; et al. Efficacy, Safety, Pharmacokinetics, and Biomarkers of Cediranib Monotherapy in Advanced Hepatocellular Carcinoma: A Phase II Study. Clin. Cancer Res. 2013, 19, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T.T.; Gerstner, E.R.; Emblem, K.E.; Duda, D.G.; Kalpathy-Cramer, J.; Snuderl, M.; Ancukiewicz, M.; Polaskova, P.; Pinho, M.C.; Jennings, D.; et al. Improved Tumor Oxygenation and Survival in Glioblastoma Patients Who Show Increased Blood Perfusion after Cediranib and Chemoradiation. Proc. Natl. Acad. Sci. USA 2013, 110, 19059–19064. [Google Scholar] [CrossRef] [Green Version]
- Weickhardt, A.J.; Williams, D.S.; Lee, C.K.; Chionh, F.; Simes, J.; Murone, C.; Wilson, K.; Parry, M.M.; Asadi, K.; Scott, A.M.; et al. Vascular Endothelial Growth Factor D Expression Is a Potential Biomarker of Bevacizumab Benefit in Colorectal Cancer. Br. J. Cancer 2015, 113, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; de Haas, S.; Kang, Y.-K.; Ohtsu, A.; Tebbutt, N.C.; Ming Xu, J.; Peng Yong, W.; Langer, B.; Delmar, P.; Scherer, S.J.; et al. Bevacizumab in Combination with Chemotherapy as First-Line Therapy in Advanced Gastric Cancer: A Biomarker Evaluation from the AVAGAST Randomized Phase III Trial. J. Clin. Oncol. 2012, 30, 2119–2127. [Google Scholar] [CrossRef]
- Benson, A.B.; Kiss, I.; Bridgewater, J.; Eskens, F.A.L.M.; Sasse, C.; Vossen, S.; Chen, J.; Van Sant, C.; Ball, H.A.; Keating, A.; et al. BATON-CRC: A Phase II Randomized Trial Comparing Tivozanib Plus MFOLFOX6 with Bevacizumab Plus MFOLFOX6 in Stage IV Metastatic Colorectal Cancer. Clin. Cancer Res. 2016, 22, 5058–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.-J.; Wei, X.; Peng, Y.; Zha, L.; Zhou, R.-B.; Shi, H.; Zhou, Q.; Liang, H.-J. Neuropilin-2 Mediates Lymphangiogenesis of Colorectal Carcinoma via a VEGFC/VEGFR3 Independent Signaling. Cancer Lett. 2015, 358, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.Q.; Lee, P.; Lin, H.; Soker, S.; Klagsbrun, M. Neuropilin-1 Expression by Tumor Cells Promotes Tumor Angiogenesis and Progression. FASEB J. 2000, 14, 2532–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Takagi, H.; Otani, A.; Koyama, S.; Kemmochi, S.; Uemura, A.; Honda, Y. Selective Induction of Neuropilin-1 by Vascular Endothelial Growth Factor (VEGF): A Mechanism Contributing to VEGF-Induced Angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.S.; Tabernero, J.; Tomášek, J.; Chau, I.; Melichar, B.; Safran, H.; Tehfe, M.A.; Filip, D.; Topuzov, E.; Schlittler, L.; et al. Biomarker Analyses in REGARD Gastric/GEJ Carcinoma Patients Treated with VEGFR2-Targeted Antibody Ramucirumab. Br. J. Cancer 2016, 115, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Bianchi, F.; Ferguson, M.; Cesario, A.; Margaritora, S.; Granone, P.; Goldstraw, P.; Tetlow, M.; Ratcliffe, C.; Nicholson, A.G.; et al. Gene Expression Signature for Angiogenic and Nonangiogenic Non-Small-Cell Lung Cancer. Oncogene 2005, 24, 1212–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.; Henriksson, R.; et al. Patients With Proneural Glioblastoma May Derive Overall Survival Benefit From the Addition of Bevacizumab to First-Line Radiotherapy and Temozolomide: Retrospective Analysis of the AVAglio Trial. J. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentink, S.; Haibe-Kains, B.; Risch, T.; Fan, J.-B.; Hirsch, M.S.; Holton, K.; Rubio, R.; April, C.; Chen, J.; Wickham-Garcia, E.; et al. Angiogenic MRNA and MicroRNA Gene Expression Signature Predicts a Novel Subtype of Serous Ovarian Cancer. PLoS ONE 2012, 7, e30269. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Motzer, R.J.; Tannir, N.M.; Porta, C.; Tomita, Y.; Maurer, M.A.; McHenry, M.B.; Rini, B.I. Efficacy of Nivolumab plus Ipilimumab According to Number of IMDC Risk Factors in CheckMate 214. Eur. Urol. 2020, 77, 449–453. [Google Scholar] [CrossRef]
- Marquardt, A.; Solimando, A.G.; Kerscher, A.; Bittrich, M.; Kalogirou, C.; Kübler, H.; Rosenwald, A.; Bargou, R.; Kollmannsberger, P.; Schilling, B.; et al. Subgroup-Independent Mapping of Renal Cell Carcinoma-Machine Learning Reveals Prognostic Mitochondrial Gene Signature Beyond Histopathologic Boundaries. Front. Oncol. 2021, 11, 621278. [Google Scholar] [CrossRef]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Novel Angiogenesis Inhibitors: Addressing the Issue of Redundancy in the Angiogenic Signaling Pathway. Cancer Treat. Rev. 2011, 37, 344–352. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A First-in-Human, First-in-Class, Phase I Study of Carlumab (CNTO 888), a Human Monoclonal Antibody against CC-Chemokine Ligand 2 in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of Angiopoietin-2 in Adaptive Tumor Resistance to VEGF Signaling Blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranjol, M.Z.I.; Zinovkin, D.A.; Maskell, A.R.T.; Stephens, L.J.; Achinovich, S.L.; Los’, D.M.; Nadyrov, E.A.; Hannemann, M.; Gutowski, N.J.; Whatmore, J.L. Cathepsin L-Induced Galectin-1 May Act as a Proangiogenic Factor in the Metastasis of High-Grade Serous Carcinoma. J. Transl. Med. 2019, 17, 216. [Google Scholar] [CrossRef] [Green Version]
- Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J. Clin. Med. 2020, 9, 675. [Google Scholar] [CrossRef]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of Tumor Invasion and Metastasis by Concurrent Inhibition of C-Met and VEGF Signaling in Pancreatic Neuroendocrine Tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Simmons, B.H.; Lee, J.H.; Lappin, P.B.; Christensen, J.G. HGF/c-Met Pathway Is One of the Mediators of Sunitinib-Induced Tumor Cell Type-Dependent Metastasis. Cancer Lett. 2012, 320, 48–55. [Google Scholar] [CrossRef]
- Desantis, V.; Frassanito, M.A.; Tamma, R.; Saltarella, I.; Di Marzo, L.; Lamanuzzi, A.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; et al. Rhu-Epo down-Regulates pro-Tumorigenic Activity of Cancer-Associated Fibroblasts in Multiple Myeloma. Ann. Hematol. 2018, 97, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Lamanuzzi, A.; Saltarella, I.; Frassanito, M.A.; Ribatti, D.; Melaccio, A.; Desantis, V.; Solimando, A.G.; Ria, R.; Vacca, A. Thrombopoietin Promotes Angiogenesis and Disease Progression in Patients with Multiple Myeloma. Am. J. Pathol. 2021, 191, 748–758. [Google Scholar] [CrossRef]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone Marrow Endothelial Cells Sustain a Tumor-Specific CD8+ T Cell Subset with Suppressive Function in Myeloma Patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef] [Green Version]
- Alard, E.; Butnariu, A.-B.; Grillo, M.; Kirkham, C.; Zinovkin, D.A.; Newnham, L.; Macciochi, J.; Pranjol, M.Z.I. Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets. Cancers 2020, 12, 1826. [Google Scholar] [CrossRef]
- Dalotto-Moreno, T.; Croci, D.O.; Cerliani, J.P.; Martinez-Allo, V.C.; Dergan-Dylon, S.; Méndez-Huergo, S.P.; Stupirski, J.C.; Mazal, D.; Osinaga, E.; Toscano, M.A.; et al. Targeting Galectin-1 Overcomes Breast Cancer-Associated Immunosuppression and Prevents Metastatic Disease. Cancer Res. 2013, 73, 1107–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, P.P. Galectin 3 as a Guardian of the Tumor Microenvironment. Biochim. Biophys. Acta 2016, 1863, 427–437. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the Hallmarks of Cancer: Galectins as Multifunctional Mediators of Tumor Progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Levicar, N.; Strojnik, T.; Kos, J.; Dewey, R.A.; Pilkington, G.J.; Lah, T.T. Lysosomal Enzymes, Cathepsins in Brain Tumour Invasion. J. Neurooncol. 2002, 58, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Vigneswaran, N.; Zacharias, W. Hypoxia Inhibits TRAIL-Induced Tumor Cell Apoptosis: Involvement of Lysosomal Cathepsins. Apoptosis 2007, 12, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.-J.; et al. Targeting Galectin-1 Inhibits Pancreatic Cancer Progression by Modulating Tumor-Stroma Crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Hosur, M.V.; Sowder, R.C.; Cachau, R.E.; Collins, J.; Silva, A.M.; Erickson, J.W. Crystal Structures of Native and Inhibited Forms of Human Cathepsin D: Implications for Lysosomal Targeting and Drug Design. Proc. Natl. Acad. Sci. USA 1993, 90, 6796–6800. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L Targeting in Cancer Treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Montemagno, C.; Pagès, G. Resistance to Anti-Angiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tumor Site | Name of the Study | Endpoint | Effect on OS Months (p-Value) | Effect on PFS Months (p-Value) |
|---|---|---|---|---|
| Prostate * | CALGB 90401 | OS | 2.4 (0.0001) | 1.1 (0.18) |
| Pancreas * | AVITA | OS | 1 (0.0002) | 1.1 (0.21) |
| Lung * | ECOG E4599 | OS | 1.7 (<0.001) | 2 (0.003) |
| Lung * | AVAIL | PFS, OS | 0.6 (0.0003) | 0.5 (0.2) |
| Kidney * | CALGB 90206 | OS | 3.3 (<0.001) | 0.9 (0.097) |
| Breast * | ECOG E2100 | PFS | 5.9 (0.001) | 1.5 (0.16) |
| Breast * | AVADO | PFS | 1.9 (0.006) | −1.7 (0.85) |
| Breast * | RIBBON-1 | PFS | 2.9 (0.0002) | 7.8 (0.27) |
| Gastric * | AVAGAST | OS | 1.4 (0.0037) | 2 (0.1) |
| Colorectal * | Hurwitz et al. (2008) | OS | 4.4 (<0.001) | 4.7 (<0.001) |
| Colorectal ** | VELOUR | OS | 2.23 (<0.001) | 1.4 (0.0032) |
| Colorectal # | RAISE | OS | 1.2 (<0.0005) | 1.6 (0.0219) |
| Colorectal ## | CORRECT | OS | 1.4 (0.0052) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribatti, D.; Solimando, A.G.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. https://doi.org/10.3390/cancers13143433
Ribatti D, Solimando AG, Pezzella F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers. 2021; 13(14):3433. https://doi.org/10.3390/cancers13143433
Chicago/Turabian StyleRibatti, Domenico, Antonio Giovanni Solimando, and Francesco Pezzella. 2021. "The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer" Cancers 13, no. 14: 3433. https://doi.org/10.3390/cancers13143433
APA StyleRibatti, D., Solimando, A. G., & Pezzella, F. (2021). The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers, 13(14), 3433. https://doi.org/10.3390/cancers13143433