
Role of Lysophospholipid Metabolism in Chronic Myelogenous Leukemia Stem Cells


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. CML Stem Cells in CML Disease
1.2. Transcriptional Control in CML Stem Cells
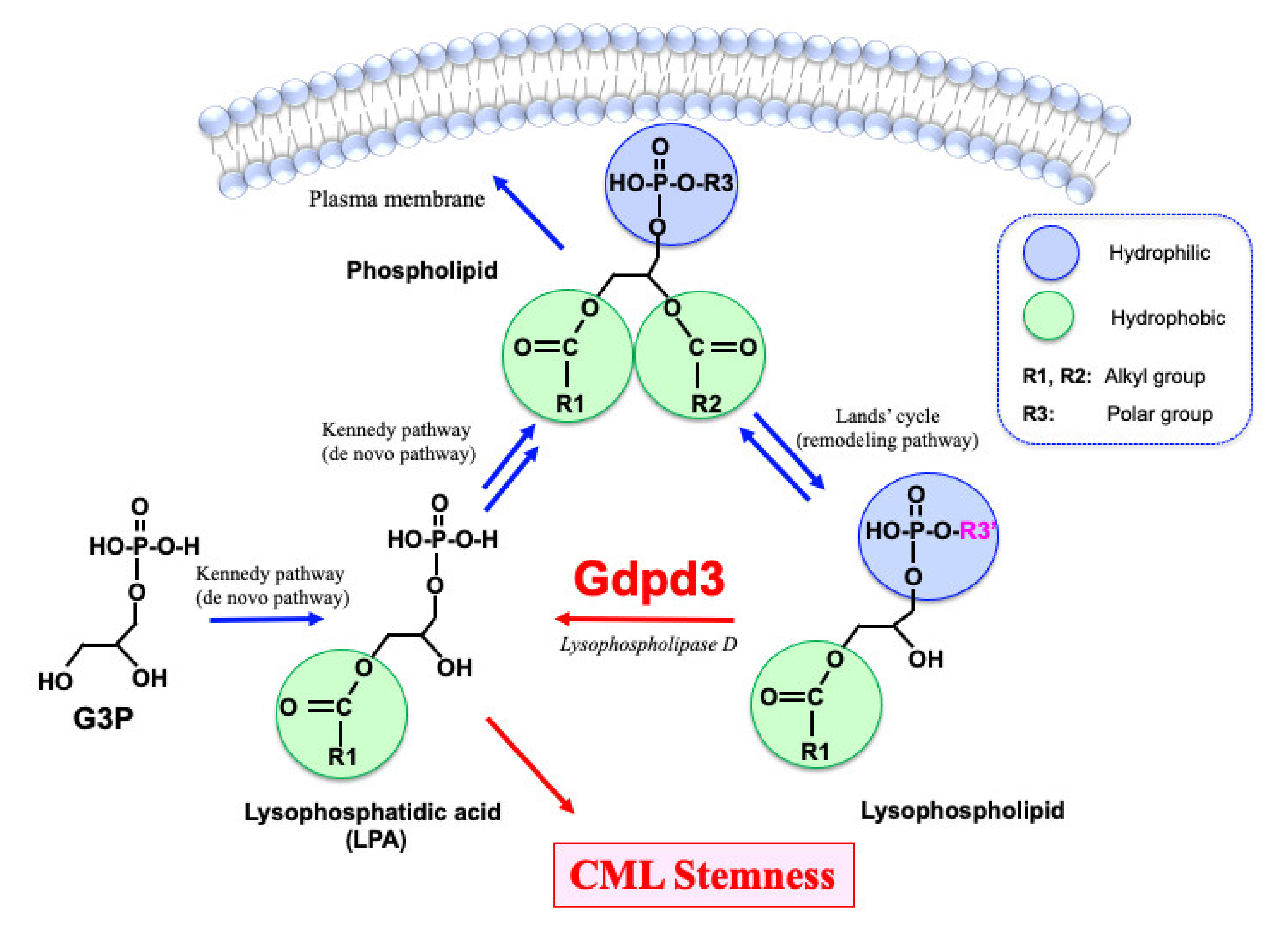
1.3. Biology of Lysophospholipids and Lysophosphatidic Acids
2. Biological Significance of Gdpd3 in CML Stem Cells
2.1. Stem Cell Quiescence and TKI Resistance
2.2. Lipidomics Analyses of the Gdpd3-Deficient CML Cells
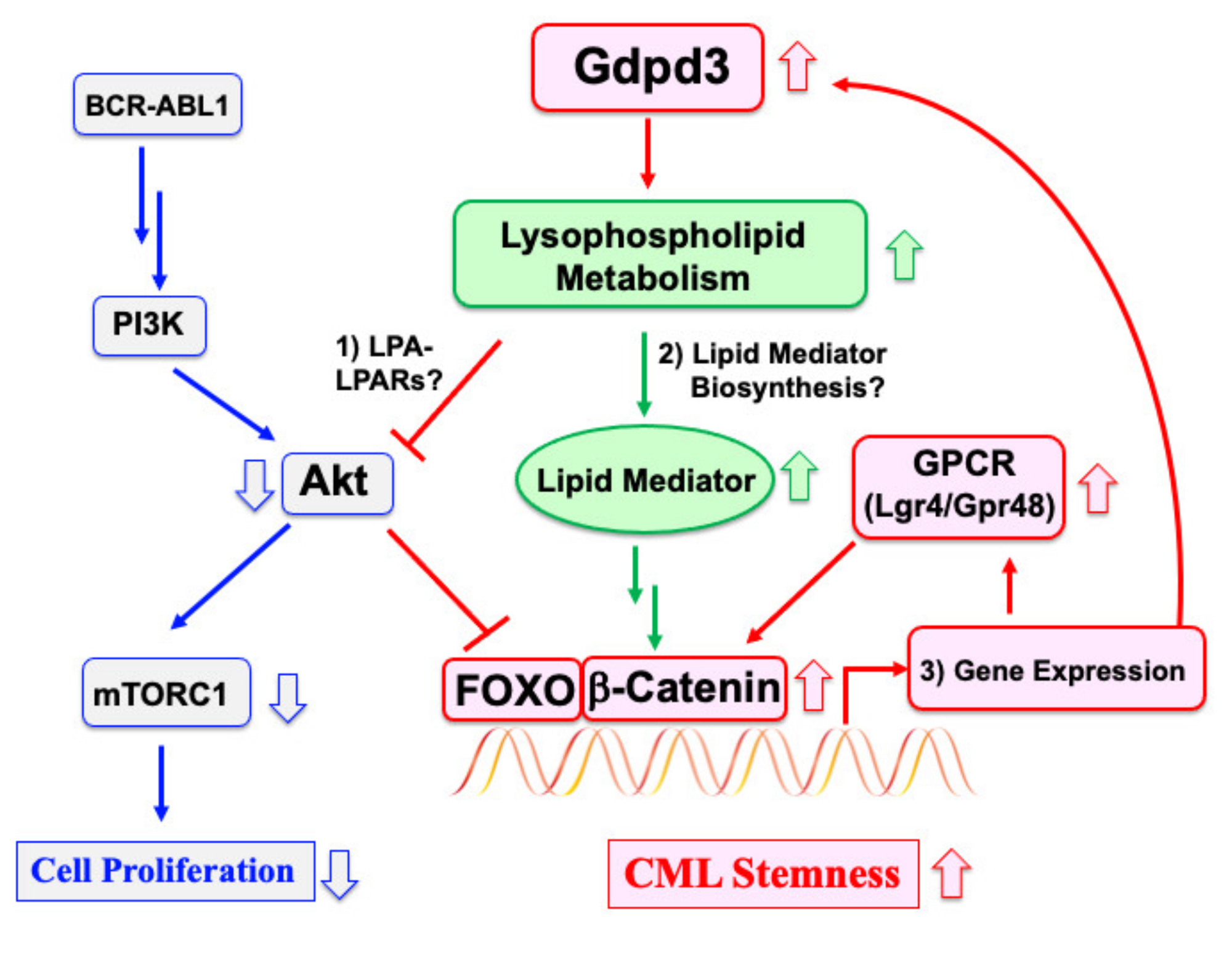
2.3. A Signaling Pathway That Regulates CML Stem Cell Quiescence
2.4. Downstream Targets Underlying CML Stemness
3. Additional Perspectives
3.1. Suppression of Akt by an LPA–LPARs Pathway
3.2. Functional Links between Lysophospholipid Metabolism and Lipid Mediator Biosynthesis
3.3. A Gene Expression Program Involving Gdpd3 and GPCRs by FOXO/β-Catenin
4. Conclusions
Funding
Conflicts of Interest
References
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Saußele, S.; Richter, J.; Hochhaus, A.; Mahon, F.X. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016, 30, 1638–1647. [Google Scholar] [CrossRef]
- Ureshino, H. Treatment-free remission and immunity in chronic myeloid leukemia. Int. J. Hematol. 2021, 113, 642–647. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.; Whetton, A.D.; et al. The anti-proliferative activity of kinase inhibitors in chronic myeloid leukaemia cells is mediated by FOXO transcription factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Ochiai, R.; Matsubara, E.; Kondo, C.; Yang, K.M.; Hoshii, T.; Araki, M.; Araki, K.; Sotomaru, Y.; Sasaki, K.; et al. The lysophospholipase D enzyme Gdpd3 is required to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 2020, 11, 4681. [Google Scholar] [CrossRef] [PubMed]
- Naka, K. New routes to eradicating chronic myelogenous leukemia stem cells by targeting metabolism. Int. J. Hematol. 2021, 113, 648–655. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Shindou, H.; Shimizu, T. Acyl-CoA: Lysophospholipid acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, N.; Kudo, T.; Yamashita, Y.; Mariggio, S.; Araki, M.; Honda, A.; Nagano, T.; Isaji, C.; Kato, N.; Corda, D.; et al. New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J. Biol. Chem. 2015, 290, 4260–4271. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.A.; Tsuboi, K.; Hussain, Z.; Yamashita, R.; Okamoto, Y.; Uyama, T.; Yamazaki, N.; Tanaka, T.; Tokumura, A.; Ueda, N. Calcium-dependent generation of N-acylethanolamines and lysophosphatidic acids by glycerophosphodiesterase GDE7. Biochim. Biophys. Acta 2016, 1861, 1881–1892. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolenaar, W.H.; Perrakis, A. Insights into autotaxin: How to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 2011, 12, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Emoto, S.; Kurano, M.; Kano, K.; Matsusaki, K.; Yamashita, H.; Nishikawa, M.; Igarashi, K.; Ikeda, H.; Aoki, J.; Kitayama, J.; et al. Analysis of glycero-lysophospholipids in gastric cancerous ascites. J. Lipid Res. 2017, 58, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Kurano, M.; Suzuki, A.; Inoue, A.; Tokuhara, Y.; Kano, K.; Matsumoto, H.; Igarashi, K.; Ohkawa, R.; Nakamura, K.; Dohi, T.; et al. Possible involvement of minor lysophospholipids in the increase in plasma lysophosphatidic acid in acute coronary syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uranbileg, B.; Ito, N.; Kurano, M.; Saigusa, D.; Saito, R.; Uruno, A.; Kano, K.; Ikeda, H.; Yamada, Y.; Sumitani, M.; et al. Alteration of the lysophosphatidic acid and its precursor lysophosphatidylcholine levels in spinal cord stenosis: A study using a rat cauda equina compression model. Sci. Rep. 2019, 9, 16578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/b-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [Green Version]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/b-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenbaum, S.P.; Ordonez-Moran, P.; Puig, I.; Chicote, I.; Arques, O.; Landolfi, S.; Fernandez, Y.; Herance, J.R.; Gispert, J.D.; Mendizabal, L.; et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012, 12, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of b-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; He, B.; Ma, X.; Yu, S.; Bhave, R.R.; Lentz, S.R.; Tan, K.; Guzman, M.L.; Zhao, C.; Xue, H.H. Prostaglandin E1 and its analog Misoprostol inhibit human CML stem cell self-renewal via EP4 receptor activation and repression of AP-1. Cell Stem Cell 2017, 21, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef]
- Igarashi, H.; Akahoshi, N.; Ohto-Nakanishi, T.; Yasuda, D.; Ishii, S. The lysophosphatidic acid receptor LPA4 regulates hematopoiesis-supporting activity of bone marrow stromal cells. Sci. Rep. 2015, 5, 11410. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Abraham, S.A.; Shan, Y.; Guo, Z.; Desouza, N.; Cheloni, G.; Li, D.; Holyoake, T.L.; Li, S. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J. Clin. Investig. 2014, 124, 3847–3862. [Google Scholar] [CrossRef] [Green Version]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARg agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Ye, X.; Hama, K.; Contos, J.J.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Paria, B.C.; Das, S.K.; Dinchuk, J.E.; Langenbach, R.; Trzaskos, J.M.; Dey, S.K. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell 1997, 91, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Salik, B.; Yi, H.; Hassan, N.; Santiappillai, N.; Vick, B.; Connerty, P.; Duly, A.; Trahair, T.; Woo, A.J.; Beck, D.; et al. Targeting RSPO3-LGR4 Signaling for Leukemia Stem Cell Eradication in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Beaulac, H.J.; Gilels, F.; Zhang, J.Y.; Jeoung, S.; White, P.A. Primed to Die: An Investigation of the Genetic Mechanisms Underlying Noise-Induced Hearing Loss and Cochlear Damage in Homozygous Foxo3-knockout Mice. Cell Death Dis. 2021, 12, 682. [Google Scholar] [CrossRef]
- Wijayatunge, R.; Holmstrom, S.R.; Foley, S.B.; Mgbemena, V.E.; Bhargava, V.; Perez, G.L.; McCrum, K.; Ross, T.S. Deficiency of the endocytic protein Hip1 leads to decreased Gdpd3 expression, low phosphocholine, and kypholordosis. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naka, K. Role of Lysophospholipid Metabolism in Chronic Myelogenous Leukemia Stem Cells. Cancers 2021, 13, 3434. https://doi.org/10.3390/cancers13143434
Naka K. Role of Lysophospholipid Metabolism in Chronic Myelogenous Leukemia Stem Cells. Cancers. 2021; 13(14):3434. https://doi.org/10.3390/cancers13143434
Chicago/Turabian StyleNaka, Kazuhito. 2021. "Role of Lysophospholipid Metabolism in Chronic Myelogenous Leukemia Stem Cells" Cancers 13, no. 14: 3434. https://doi.org/10.3390/cancers13143434
APA StyleNaka, K. (2021). Role of Lysophospholipid Metabolism in Chronic Myelogenous Leukemia Stem Cells. Cancers, 13(14), 3434. https://doi.org/10.3390/cancers13143434