
DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model


Abstract
:Simple Summary
Abstract
1. Introduction
2. DNA Methylation Functions in Hematopoiesis
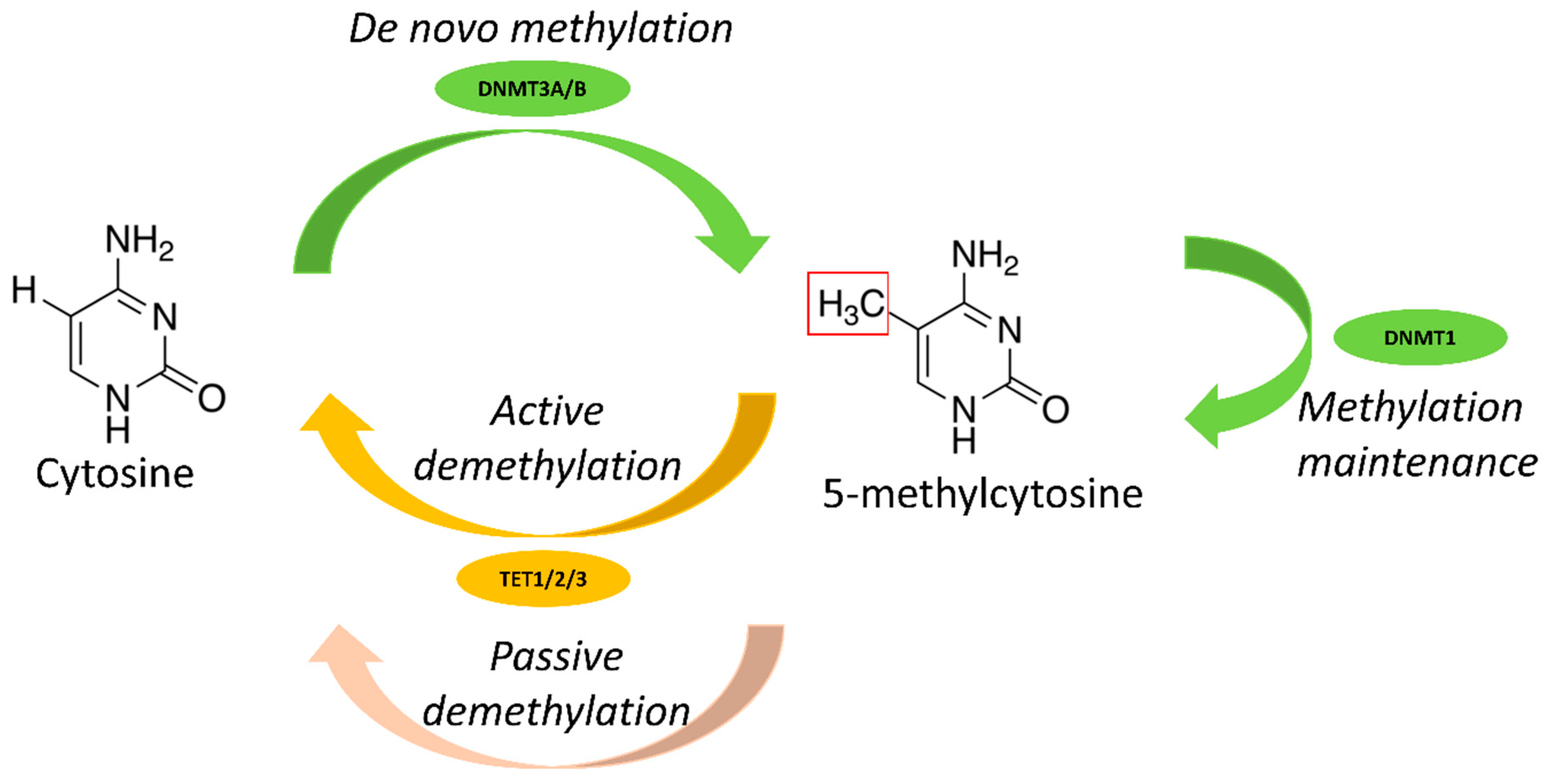
2.1. DNA Methylation
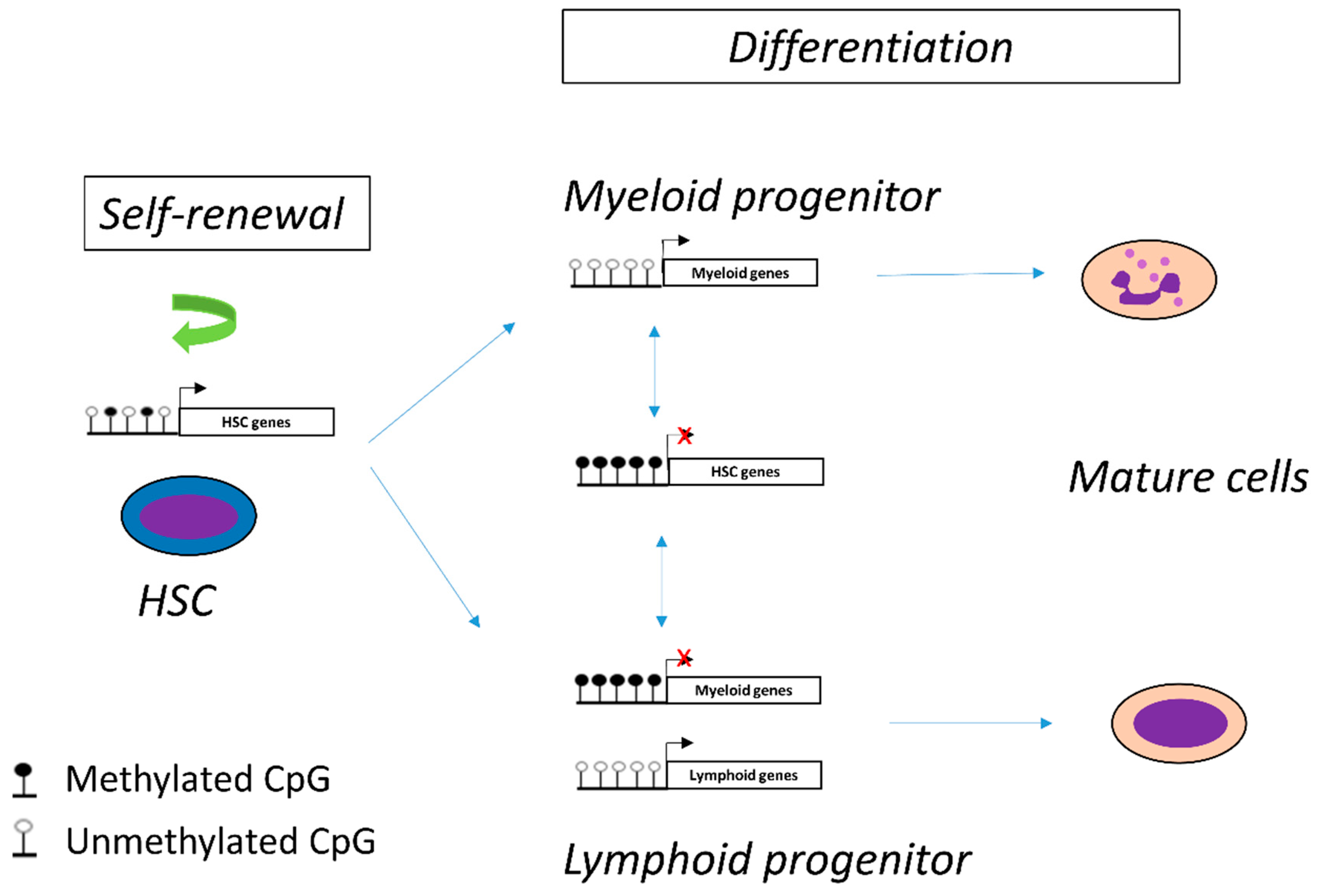
2.2. DNA Methylation in Hematopoiesis
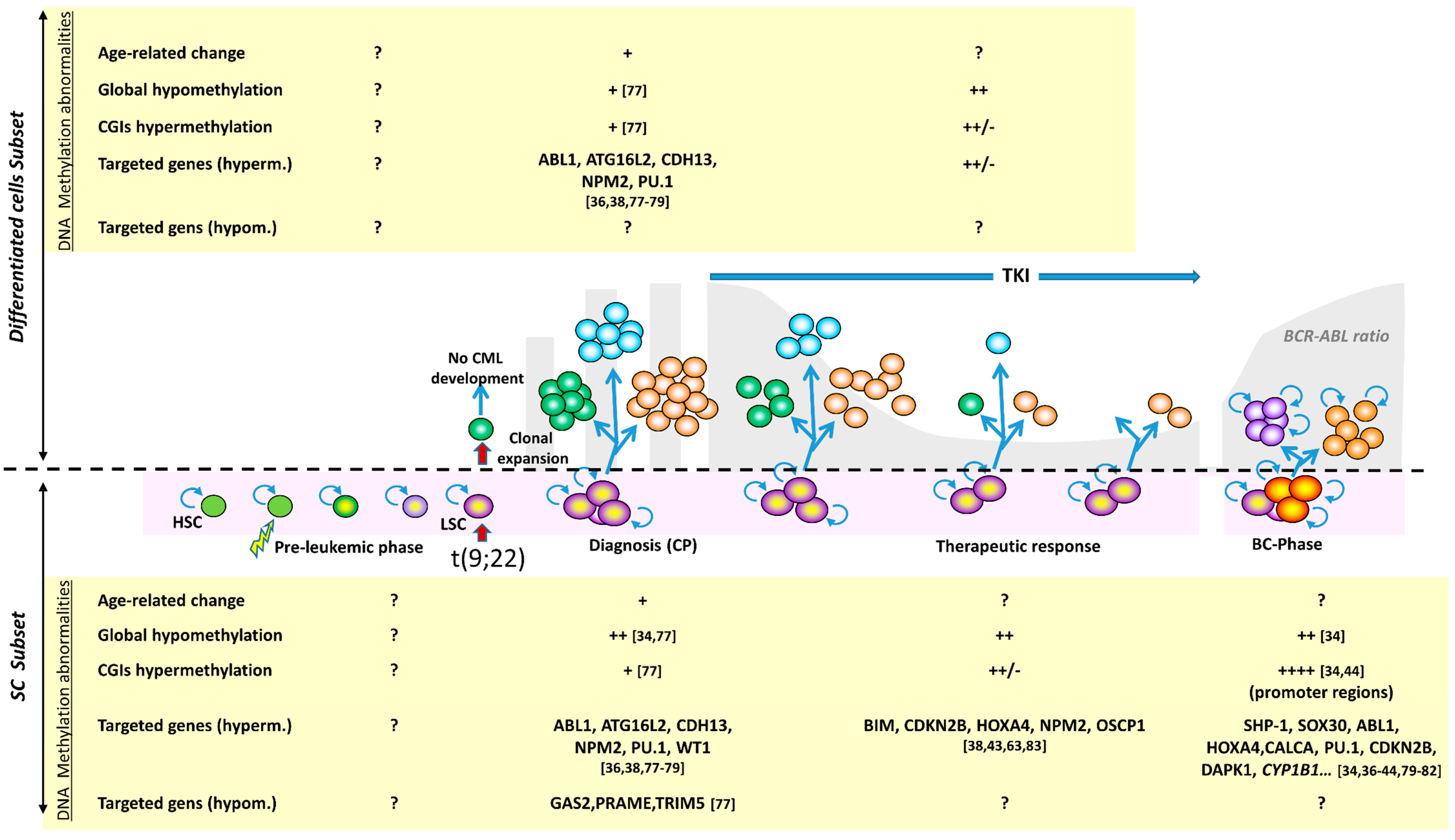
3. DNA Methylation Abnormalities in CML
3.1. DNA Methylation Abnormalities of BC-CML Cells
3.1.1. Methylation Abnormalities Are Associated with CML Progression
3.1.2. Differences and Similarities with Ph1-Negative Acute Myeloid Leukemia (AML)
3.1.3. Epigenetics, CML and AML Predisposition
3.2. DNA Methylation in CP-CML and Intra-Clonal Heterogeneity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Investigated | TET1 | TET2 | TET3 | DNMT3A | DNMT1 DNMT3B | ASXL1 | Epigenetic Regulator | DNA Methylation |
|---|---|---|---|---|---|---|---|---|
| 4 genes [84] | / | 1/91 | / | / | / | 8/91 | 9/91 | 1/91 |
| 25 genes [76] | / | 1/15 | / | 2/15 | / | 1/15 | 4/15 | 3/15 |
| 71 genes [85] | 0/124 | 1/124 | / | 4/124 | 0/124 | 9/124 | 37/124 | 5/124 |
| 92 genes [62] | 0/100 | 6/100 | / | 2/100 | 0/100 | 9/100 | 19/100 | 8/100 |
| Whole exome [86] | 0/24 | 1/24 | 1/24 | 0/24 | 0/24 | 3/24 | 6/24 | 2/24 |
| Whole exome [87] | 0/40 | 1/40 | 0/40 | 1/40 | 0/40 | 6/40 | 10/40 | 2/40 |
| Whole exome [32] | 0/46 | 0/46 | 0/46 | 0/46 | 0/46 | 9/46 | 11/46 | 1/46 |
| Total | 0/334 | 11/440 | 1/110 | 9/349 | 0/334 | 45/440 | 96/440 | 22/440 |
| % | 0% | 2.50% | 0.90% | 2.60% | 0% | 10.20% | 21.80% | 5% |
4. Pathophysiology of DNA Methylation Changes
5. Perspectives
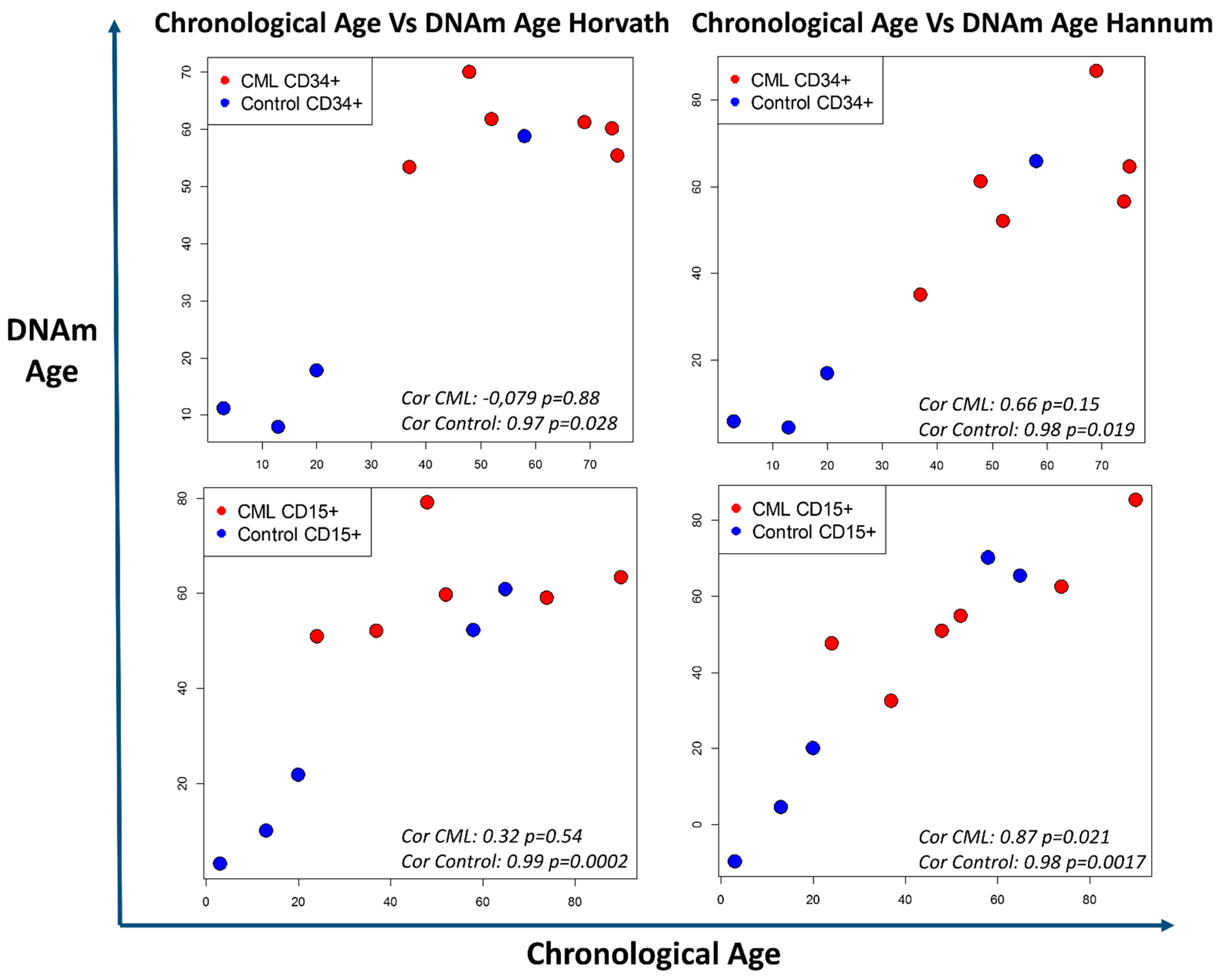
5.1. DNA Methylation, Biological Age, and CML Predisposition
5.2. DNA Methylation as a Therapeutic Target
5.3. DNA Methylation as a Biomarker of TKI Resistance and Intra-Clonal Heterogeneity
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hoffmann, V.S.; Baccarani, M.; Hasford, J.; Lindoerfer, D.; Burgstaller, S.; Sertic, D.; Costeas, P.; Mayer, J.; Indrak, K.; Everaus, H.; et al. The EUTOS population-based registry: Incidence and clinical characteristics of 2904 CML patients in 20 European Countries. Leukemia 2015, 29, 1336–1343. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nat. Cell Biol. 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Giralt, S.; Kantarjian, H.; Talpaz, M. The natural history of chronic myelogenous leukemia in the interferon era. Semin. Hematol. 1995, 32, 152–158. [Google Scholar]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne, G.; Guilhot, J.; Rea, D.; Rigal-Huguet, F.; Nicolini, F.; Charbonnier, A.; Guerci-Bresler, A.; Legros, L.; Varet, B.; Gardembas, M.; et al. Long-Term Follow-Up of the French Stop Imatinib (STIM1) Study in Patients With Chronic Myeloid Leukemia. J. Clin. Oncol. 2017, 35, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Campiotti, L.; Suter, M.B.; Guasti, L.; Piazza, R.; Gambacorti-Passerini, C.; Grandi, A.M.; Squizzato, A. Imatinib discontinuation in chronic myeloid leukaemia patients with undetectable BCR-ABL transcript level: A systematic review and a meta-analysis. Eur. J. Cancer 2017, 77, 48–56. [Google Scholar] [CrossRef]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.J.W.M.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.-H.; Ame, S.; et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef] [Green Version]
- Turhan, A.G.; Hugues, P.; Sorel, N.; Desterke, C.; Bourhis, J.-H.; Bennaceur-Griscelli, A.; Chomel, J.C. Evidence of BCR-ABL1-positive progenitor spread in blood during molecular recurrence after TKI discontinuation in chronic myeloid leukemia (CML). Leuk. Lymphoma 2020, 61, 1719–1723. [Google Scholar] [CrossRef]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef] [Green Version]
- Koschmieder, S.; Vetrie, D. Epigenetic dysregulation in chronic myeloid leukaemia: A myriad of mechanisms and therapeutic options. Semin. Cancer Biol. 2018, 51, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Polakova, K.M.; Koblihova, J.; Stopka, T. Role of Epigenetics in Chronic Myeloid Leukemia. Curr. Hematol. Malign. Rep. 2013, 8, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nat. Cell Biol. 2009, 463, 1042–1047. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA Glycosylase Is Essential for Active DNA Demethylation by Linked Deamination-Base Excision Repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reik, W.; Collick, A.; Norris, M.L.; Barton, S.C.; Surani, M.A. Genomic imprinting determines methylation of parental alleles in transgenic mice. Nat. Cell Biol. 1987, 328, 248–251. [Google Scholar] [CrossRef]
- Riggs, A. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nat. Cell Biol. 2010, 467, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Bocker, M.T.; Hellwig, I.; Breiling, A.; Eckstein, V.; Ho, A.D.; Lyko, F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 2011, 117, e182–e189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farlik, M.; Halbritter, F.; Müller, F.; Choudry, F.A.; Ebert, P.; Klughammer, J.; Farrow, S.; Santoro, A.; Ciaurro, V.; Mathur, A.; et al. DNA Methylation Dynamics of Human Hematopoietic Stem Cell Differentiation. Cell Stem Cell 2016, 19, 808–822. [Google Scholar] [CrossRef] [Green Version]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- A Burke, B.; Carroll, M. BCR–ABL: A multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia 2010, 24, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef]
- Grossmann, V.; Kohlmann, A.; Zenger, M.; Schindela, S.; Eder, C.; Weissmann, S.; Schnittger, S.; Kern, W.; Muller, M.C.; Hochhaus, A.; et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 2011, 25, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Javed, A.; Lee, K.L.; Pathiraja, T.N.; Liu, X.; Malik, S.; Soh, S.X.; Heng, X.T.; Takahashi, N.; Tan, J.H.J.; et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood 2020, 135, 2337–2353. [Google Scholar] [CrossRef]
- Johansson, B.; Fioretos, T.; Mitelman, F. Cytogenetic and Molecular Genetic Evolution of Chronic Myeloid Leukemia. Acta Haematol. 2002, 107, 76–94. [Google Scholar] [CrossRef]
- Dunwell, T.; Hesson, L.; A Rauch, T.; Wang, L.; E Clark, R.; Dallol, A.; Gentle, D.; Catchpoole, D.; Maher, E.R.; Pfeifer, G.P.; et al. A Genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol. Cancer 2010, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.J.; Denkers, F.; Valk, P.; Cornelissen, J.J.; Schuurhuis, G.-J.; Ossenkoppele, G.J. Methylation patterns in CD34 positive chronic myeloid leukemia blast crisis cells. Haematologica 2010, 95, 1036–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, J.; Gharibyan, V.; Estecio, M.; Kondo, K.; He, R.; Chung, W.; Lu, Y.; Zhang, N.; Liang, S.; Kantarjian, H.M.; et al. Aberrant DNA Methylation Is Associated with Disease Progression, Resistance to Imatinib and Shortened Survival in Chronic Myelogenous Leukemia. PLoS ONE 2011, 6, e22110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-J.; Wen, X.-M.; Zhou, J.-D.; Gu, Y.; Xu, Z.-J.; Guo, H.; Ma, J.-C.; Yuan, Q.; Chen, Q.; Lin, J.; et al. SOX30 methylation correlates with disease progression in patients with chronic myeloid leukemia. OncoTargets Ther. 2019, 12, 4789–4794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strathdee, G.; Holyoake, T.; Sim, A.; Parker, A.; Oscier, D.G.; Melo, J.V.; Meyer, S.; Eden, T.; Dickinson, A.M.; Mountford, J.; et al. Inactivation of HOXA Genes by Hypermethylation in Myeloid and Lymphoid Malignancy is Frequent and Associated with Poor Prognosis. Clin. Cancer Res. 2007, 13, 5048–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Wang, Y.-L.; Lin, J.; Yao, D.-M.; Xu, W.-R.; Wu, C.-Y. Aberrant methylation of the death-associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur. J. Haematol. 2009, 82, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, L.; Pan, Y.; Yang, J.; Shang, Y.; Luo, J. Methylation and decreased expression of SHP-1 are related to disease progression in chronic myelogenous leukemia. Oncol. Rep. 2014, 31, 2438–2446. [Google Scholar] [CrossRef] [Green Version]
- Behzad, M.M.; Shahrabi, S.; Jaseb, K.; Bertacchini, J.; Ketabchi, N.; Saki, N. Aberrant DNA Methylation in Chronic Myeloid Leukemia: Cell Fate Control, Prognosis, and Therapeutic Response. Biochem. Genet. 2018, 56, 149–175. [Google Scholar] [CrossRef]
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C.; et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Court, F.; Le Boiteux, E.; Fogli, A.; Müller-Barthélémy, M.; Vaurs-Barrière, C.; Chautard, E.; Pereira, B.; Biau, J.; Kemeny, J.-L.; Khalil, T.; et al. Transcriptional alterations in glioma result primarily from DNA methylation–independent mechanisms. Genome Res. 2019, 29, 1605–1621. [Google Scholar] [CrossRef] [Green Version]
- Akalin, A.; Garrett-Bakelman, F.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.; Huang, Y.; Biswas, R.; Hess, J.; et al. Base-Pair Resolution DNA Methylation Sequencing Reveals Profoundly Divergent Epigenetic Landscapes in Acute Myeloid Leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Schoofs, T.; Berdel, W.E.; Mullertidow, C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 2014, 28, 1–14. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; E Figueroa, M.; Sinha, A.U.; Stubbs, M.C.; Feng, Z.; Valk, P.J.M.; Delwel, R.; Döhner, K.; Bullinger, L.; Kung, A.; et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef]
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.; Lind, E.F.; Brenner, D.; Bruestle, A.; Harris, I.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nat. Cell Biol. 2012, 488, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.-J.; Xu, J.; Gu, Z.-H.; Pan, C.-M.; Lu, G.; Shen, Y.; Shi, J.-Y.; Zhu, Y.-M.; Tang, L.; Zhang, X.-W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.; Larson, D.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaanderen, J.; Lan, Q.; Kromhout, H.; Rothman, N.; Vermeulen, R. Occupational benzene exposure and the risk of chronic myeloid leukemia: A meta-analysis of cohort studies incorporating study quality dimensions. Am. J. Ind. Med. 2012, 55, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Health 2010, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Cortes, J.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J.; et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Hills, R.; Moorman, A.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.; Burnett, A.K. National Cancer Research Institute Adult Leukaemia Working Group Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Feurstein, S.; Drazer, M.W.; Godley, L.A. Genetic predisposition to leukemia and other hematologic malignancies. Semin. Oncol. 2016, 43, 598–608. [Google Scholar] [CrossRef]
- Ernst, T.; Busch, M.; Rinke, J.; Ernst, J.; Haferlach, C.; Beck, J.F.; Hochhaus, A.; Gruhn, B. Frequent ASXL1 mutations in children and young adults with chronic myeloid leukemia. Leukemia 2018, 32, 2046–2049. [Google Scholar] [CrossRef]
- Kim, T.; Tyndel, M.S.; Kim, H.J.; Ahn, J.-S.; Choi, S.H.; Park, H.J.; Kim, Y.-K.; Kim, S.Y.; Lipton, J.H.; Zhang, Z.; et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood 2017, 129, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, M.H.; Baba, A.A.; Husin, A.; Sulong, S.; Hassan, R.; Sim, G.A.; Wahid, S.F.A.; Ankathil, R. HOXA4Gene Promoter Hypermethylation as an Epigenetic Mechanism Mediating Resistance to Imatinib Mesylate in Chronic Myeloid Leukemia Patients. BioMed Res. Int. 2012, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rafei, H.; DiNardo, C.D. Hereditary myeloid malignancies. Best Pr. Res. Clin. Haematol. 2019, 32, 163–176. [Google Scholar] [CrossRef]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Cohen, N.M.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Trinchant, N.M.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, F.; Lee, S.C.; Poran, A.; Chaligne, R.; Gaiti, F.; Gross, B.; Murali, R.R.; Deochand, S.D.; Ang, C.; Jones, P.W.; et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020, 52, 378–387. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nat. Cell Biol. 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rinke, J.; Schäfer, V.; Schnittger, S.; Kohlmann, A.; Obstfelder, E.; Kunert, C.; Ziermann, J.; Winkelmann, N.; Eigendorff, E.; et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia 2014, 28, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Maupetit-Méhouas, S.; Court, F.; Bourgne, C.; Guerci-Bresler, A.; Cony-Makhoul, P.; Johnson, H.; Etienne, G.; Rousselot, P.; Guyotat, D.; Janel, A.; et al. DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34+ CD15− cells in early chronic-phase chronic myeloid leukemia. Mol. Oncol. 2018, 12, 814–829. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Jiang, G.; Zaydan, M.A.; La Russa, V.F.; Safah, H.; Ehrlich, M. ABL1 promoter methylation can exist independently of BCR-ABL transcription in chronic myeloid leukemia hematopoietic progenitors. Cancer Res. 2001, 61, 6931–6937. [Google Scholar] [PubMed]
- Yang, H.; Liang, H.; Yan, J.-S.; Tao, R.; Hao, S.-G.; Ma, L.-Y. Down-regulation of hematopoiesis master regulator PU.1 via aberrant methylation in chronic myeloid leukemia. Int. J. Hematol. 2012, 96, 65–73. [Google Scholar] [CrossRef]
- Zion, M.; Ben-Yehuda, D.; Avraham, A.; Cohen, O.; Wetzler, M.; Melloul, D.; Ben-Neriah, Y. Progressive de novo DNA methylation at the bcr-abl locus in the course of chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 1994, 91, 10722–10726. [Google Scholar] [CrossRef] [Green Version]
- Annamaneni, S.; Kagita, S.; Gorre, M.; Digumarti, R.R.; Satti, V.; Battini, M.R. Methylation status ofCEBPAgene promoter in chronic myeloid leukemia. Hematology 2013, 19, 42–44. [Google Scholar] [CrossRef]
- Nelkin, B.D.; Przepiorka, D.; Burke, P.J.; Thomas, E.D.; Baylin, S.B. Abnormal Methylation of the Calcitonin Gene Marks Progression of Chronic Myelogenous Leukemia. Blood 1991, 77, 2431–2434. [Google Scholar] [CrossRef] [Green Version]
- José-Eneriz, E.S.; Agirre, X.; Jiménez-Velasco, A.; Cordeu, L.; Martín, V.; Arqueros, V.; Gárate, L.; Fresquet, V.; Cervantes, F.; Martinez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 2009, 45, 1877–1889. [Google Scholar] [CrossRef]
- Roche-Lestienne, C.; Fi-LMC Group; Marceau, A.; Labis, E.; Nibourel, O.; Coiteux, V.; Guilhot, J.; Legros, L.; Nicolini, F.; Rousselot, P.; et al. Mutation analysis of TET2, IDH1, IDH2 and ASXL1 in chronic myeloid leukemia. Leukemia 2011, 25, 1661–1664. [Google Scholar] [CrossRef] [Green Version]
- Nteliopoulos, G.; Bazeos, A.; Claudiani, S.; Gerrard, G.; Curry, E.; Szydlo, R.; Alikian, M.; Foong, H.E.; Nikolakopoulou, Z.; Loaiza, S.; et al. Somatic variants in epigenetic modifiers can predict failure of response to imatinib but not to second-generation tyrosine kinase inhibitors. Haematologica 2019, 104, 2400–2409. [Google Scholar] [CrossRef] [Green Version]
- Togasaki, E.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Takeuchi, M.; Oshima, M.; Saraya, A.; Iwama, A.; Yokote, K.; Sakaida, E.; et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017, 7, e559. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.A.; Kankainen, M.; Ojala, T.; Koskenvesa, P.; Eldfors, S.; Ghimire, B.; Kumar, A.; Kytölä, S.; Kamel, M.M.; Heckman, C.A.; et al. Mutation accumulation in cancer genes relates to nonoptimal outcome in chronic myeloid leukemia. Blood Adv. 2020, 4, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Hjort, E.E.; Huang, W.; Hu, L.; Eklund, E.A. Bcr-abl regulates Stat5 through Shp2, the interferon consensus sequence binding protein (Icsbp/Irf8), growth arrest specific 2 (Gas2) and calpain. Oncotarget 2016, 7, 77635–77650. [Google Scholar] [CrossRef] [Green Version]
- Schenk, T.; Stengel, S.; Goellner, S.; Steinbach, D.; Saluz, H.P. Hypomethylation ofPRAMEis responsible for its aberrant overexpression in human malignancies. Genes Chromosom. Cancer 2007, 46, 796–804. [Google Scholar] [CrossRef]
- Rosenfeld, C.; Cheever, M.A.; Gaiger, A. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: Therapeutic potential of WT1 targeted therapies. Leukemia 2003, 17, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; Shah, J.S.; Dhungel, B.P.; Monteuuis, G.; Luu, P.-L.; Petrova, V.; Metierre, C.; Nair, S.S.; Bailey, C.G.; Saunders, V.A.; et al. Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients. Cancers 2020, 12, 3738. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Jankowska, A.M.; McDevitt, M.A.; O’Keefe, C.; Dujardin, S.; Cazzolli, H.; Przychodzen, B.; Prince, C.; Nicoll, J.; Siddaiah, H.; et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 2011, 117, e198–e206. [Google Scholar] [CrossRef] [PubMed]
- Amabile, G.; Di Ruscio, A.; Muller, F.; Welner, R.S.; Yang, H.; Ebralidze, A.K.; Zhang, H.; Levantini, E.; Qi, L.; Martinelli, G.; et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Dueñas, C.; Gonzalez-Herrero, I.; Sehgal, L.; Garcia-Ramirez, I.; Rodríguez-Hernández, G.; Pintado, B.; Blanco, O.; Criado, F.J.G.; Cenador, M.B.G.; Green, M.R.; et al. Dnmt1 links BCR-ABLp210 to epigenetic tumor stem cell priming in myeloid leukemia. Leukemia 2018, 33, 249–278. [Google Scholar] [CrossRef]
- Mahon, F.X.; Deininger, M.W.; Schultheis, B.; Chabrol, J.; Reiffers, J.; Goldman, J.M.; Melo, J.V. Selection and Characterization of BCR-ABL Positive Cell Lines with Differential Sensitivity to the Tyrosine Kinase Inhibitor STI571: Diverse Mechanisms of Resistance. Blood 2000, 96, 1070–1079. [Google Scholar] [CrossRef]
- Barnes, D.J.; Schultheis, B.; Adedeji, S.; Melo, J.V. Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene 2005, 24, 6432–6440. [Google Scholar] [CrossRef]
- Bugler, J.; Kinstrie, R.; Scott, M.; Vetrie, D. Epigenetic Reprogramming and Emerging Epigenetic Therapies in CML. Front. Cell Dev. Biol. 2019, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, I.; Mori, M.; Yamasaki, I.; Daibata, M. Is There an Entity of Radiation-Induced Chronic Myeloid Leukemia? Report of a Case and Brief Review of the Literature. J. Clin. Exp. Hematop. 2020, 60, 24–25. [Google Scholar] [CrossRef]
- Radivoyevitch, T.; Hlatky, L.; Landaw, J.; Sachs, R.K. Quantitative modeling of chronic myeloid leukemia: Insights from radiobiology. Blood 2012, 119, 4363–4371. [Google Scholar] [CrossRef] [Green Version]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, 107–121. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.-C.; Roetker, N.S.; Just, A.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Vallerga, C.L.; Walker, R.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.; Gao, Y.; Wang, J.; Li, X.; Shang, S.; Wang, Y.; Guo, S.; Zhou, H.; Liu, H.; Sun, D.; et al. CancerClock: A DNA Methylation Age Predictor to Identify and Characterize Aging Clock in Pan-Cancer. Front. Bioeng. Biotechnol. 2019, 7, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortorella, S.M.; Hung, A.; Karagiannis, T.C. The Implication of Cancer Progenitor Cells and the Role of Epigenetics in the Development of Novel Therapeutic Strategies for Chronic Myeloid Leukemia. Antioxid. Redox Signal. 2015, 22, 1425–1462. [Google Scholar] [CrossRef]
- Skórski, T. BCR/ABL regulates response to DNA damage: The role in resistance to genotoxic treatment and in genomic instability. Oncogene 2002, 21, 8591–8604. [Google Scholar] [CrossRef] [Green Version]
- Fabianowska-Majewska, K.; Wyczechowska, D.; Czyz, M. Inhibition of dna methylation by 5-aza-2’-deoxycytidine correlates with induction of K562 cells differentiation. Adv. Exp. Med. Biol. 2000, 486, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.-P.J.; Gharibyan, V.; Cortes, J.; Jelinek, J.; Morris, G.; Verstovsek, S.; Talpaz, M.; Garcia-Manero, G.; Kantarjian, H.M. Phase II Study of Low-Dose Decitabine in Patients With Chronic Myelogenous Leukemia Resistant to Imatinib Mesylate. J. Clin. Oncol. 2005, 23, 3948–3956. [Google Scholar] [CrossRef]
- Oki, Y.; Kantarjian, H.M.; Gharibyan, V.; Jones, D.; O’Brien, S.; Srdan, V.; Cortes, J.; Morris, G.M.; Garcia-Manero, G.; Issa, J.-P. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer 2007, 109, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Cortes, J.; Giles, F.J.; Faderl, S.; Issa, J.-P.; Garcia-Manero, G.; Rios, M.B.; Shan, J.; Andreeff, M.; et al. Results of decitabine (5-aza-2′deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer 2003, 98, 522–528. [Google Scholar] [CrossRef]
- Abaza, Y.; Kantarjian, H.; Alwash, Y.; Borthakur, G.; Champlin, R.; Kadia, T.; Garcia-Manero, G.; Daver, N.; Ravandi, F.; Srdan, V.; et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am. J. Hematol. 2020, 95, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Chandhok, N.S.; Prebet, T. Insights into novel emerging epigenetic drugs in myeloid malignancies. Ther. Adv. Hematol. 2019, 10, 2040620719866081. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Santini, V.; Almeida, A.; Platzbecker, U.; Jonasova, A.; Silverman, L.R.; Falantes, J.; Reda, G.; Buccisano, F.; Fenaux, P.; et al. Phase III, Randomized, Placebo-Controlled Trial of CC-486 (Oral Azacitidine) in Patients with Lower-Risk Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1426–1436. [Google Scholar] [CrossRef]
- Kulis, M.; Heath, S.C.; Bibikova, M.; Queiros, A.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Oakes, C.C.; Claus, R.; Gu, L.; Assenov, Y.; Hüllein, J.; Zucknick, M.; Bieg, M.; Brocks, D.; Bogatyrova, O.; Schmidt, C.R.; et al. Evolution of DNA Methylation Is Linked to Genetic Aberrations in Chronic Lymphocytic Leukemia. Cancer Discov. 2014, 4, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, C.C.; Seifert, M.; Assenov, Y.; Gu, L.; Przekopowitz, M.; Ruppert, A.S.; Wang, Q.; Imbusch, C.D.; Serva, A.; Koser, S.D.; et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 2016, 48, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Wierzbinska, J.A.; Plass, C.; Rosenquist, R. Epigenetic deregulation in chronic lymphocytic leukemia: Clinical and biological impact. Semin. Cancer Biol. 2018, 51, 1–11. [Google Scholar] [CrossRef]
- Queiros, A.; Villamor, N.; Clot, G.; Martineztrillos, A.; Kulis, M.; Navarro, A.; Penas, E.M.M.; Jayne, S.; Majid, A.M.S.A.; Richter, J.A.; et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2014, 29, 598–605. [Google Scholar] [CrossRef] [PubMed]




| DNA Methylation Alterations | Observations | Main Contribution |
|---|---|---|
| Genetic driver: BCR-ABL1 | BCR-ABL1 induces DNA hypomethylation through DNMT1 overexpression in mice [95] BCR-ABL1 induces an aberrant DNA methylation profile that is reversed upon BCR-ABL1 repression in mice [94] Disappearance of DNA methylation abnormalities at remission after TKI treatment in patients with CP-CML [92] | Impact of BCR-ABL on DNA methylation of the CML clone |
| Inter-individual variability | DNA methylation variation profile in different samples at diagnosis [34,44,77] | Inter-patient variability of DNA methylation suggesting BCR-ABL- independent mechanisms |
| Influence of cell origin at BC transformation | Myeloid and lymphoid blast phase cells have similar methylation profiles [34] | DNA methylation modification is more dependent on LSCs than cell lineage commitment |
| Mutations in DNA methylation regulators | 5% in CP-CML [32,62,76,84,85,86,87] 16% of AP/BC-CML [51] | Mutations in DNA methylation regulators are found in a minority of patients and in a fraction of the CML clone |
| Differentiated vs immature CML cells | DNA methylation alterations specific of immature and mature cells [34,77] | Intra-clonal heterogeneity of DNA methylation Possible influence of stem cell status on DNA methylation |
| Polycomb complex (EZH2) | Implication in transformation to the blast phase through DNA hypermethylation [34] | EZH2 is involved in CML aggressiveness through DNA methylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebecque, B.; Bourgne, C.; Vidal, V.; Berger, M.G. DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model. Cancers 2021, 13, 3587. https://doi.org/10.3390/cancers13143587
Lebecque B, Bourgne C, Vidal V, Berger MG. DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model. Cancers. 2021; 13(14):3587. https://doi.org/10.3390/cancers13143587
Chicago/Turabian StyleLebecque, Benjamin, Céline Bourgne, Véronique Vidal, and Marc G. Berger. 2021. "DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model" Cancers 13, no. 14: 3587. https://doi.org/10.3390/cancers13143587
APA StyleLebecque, B., Bourgne, C., Vidal, V., & Berger, M. G. (2021). DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model. Cancers, 13(14), 3587. https://doi.org/10.3390/cancers13143587