
Targeting Immune Modulators in Glioma While Avoiding Autoimmune Conditions

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
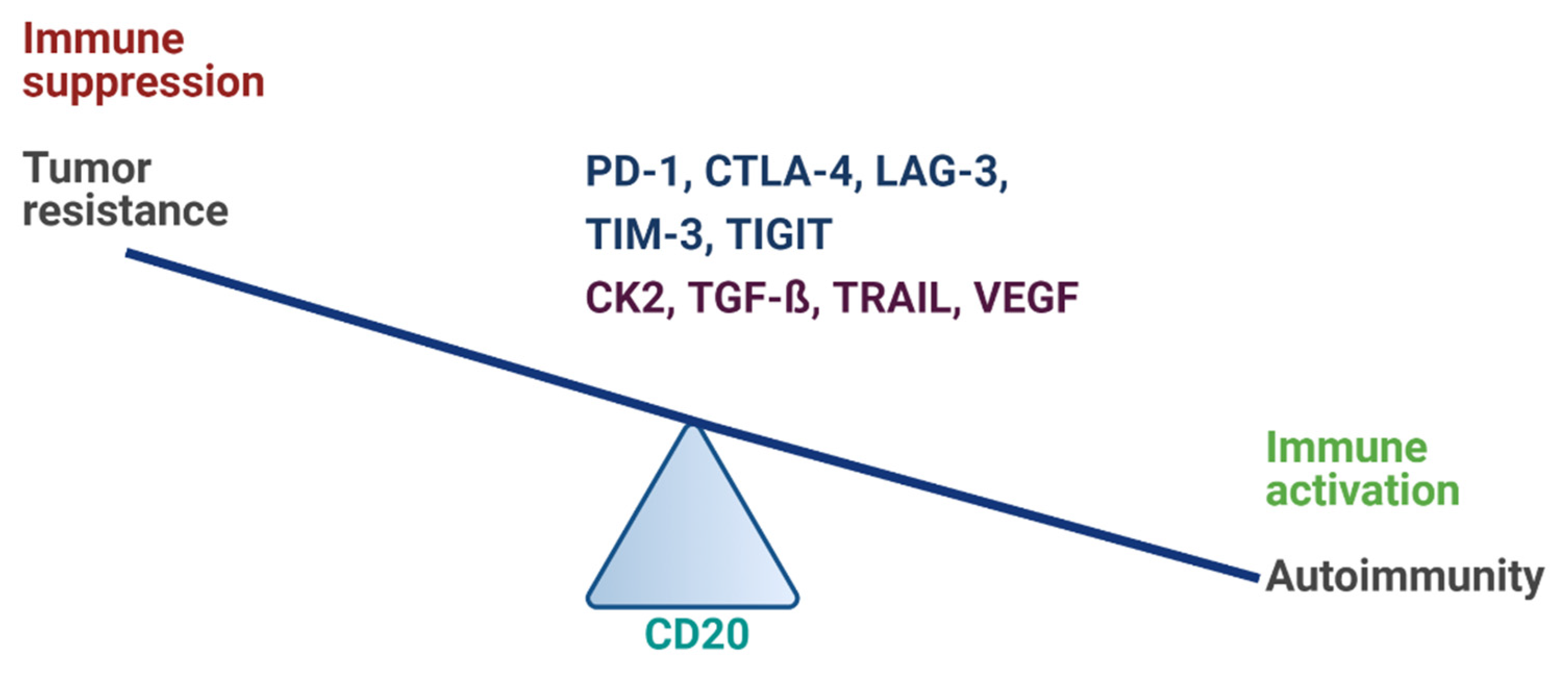
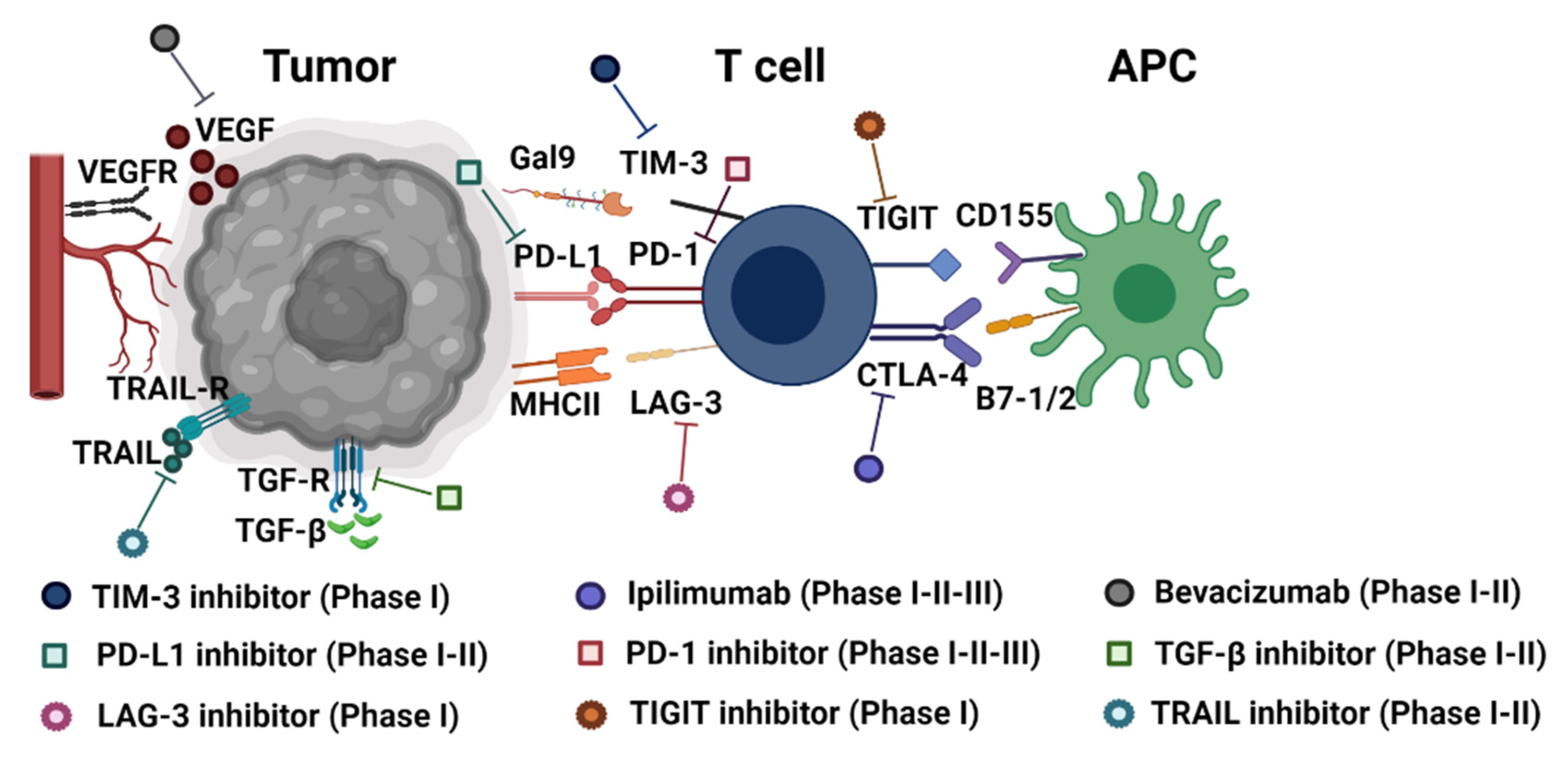
2. Immune Checkpoints Overview
3. Classical Immune Checkpoint Molecules: Efficiency and Limitations
3.1. PD-1 (CD279)
3.2. CTLA-4 (CD152)
3.3. LAG-3 (CD223)
3.4. TIM-3 (CD366)
3.5. TIGIT (Vstm3, WUCAM)
4. Pathogenic Infiltrating Th17 Cells
5. Nonclassical Immune Modulators
5.1. CK2
5.2. TGF-β
5.3. TRAIL (CD253)
5.4. VEGF
5.5. CD20
6. Combination Therapies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steinman, L. Elaborate interactions between the immune and nervous systems. Nat. Immunol. 2004, 5, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.; Loane, D.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Buscemi, L.; Price, M.; Bezzi, P.; Hirt, L. Spatio-temporal overview of neuroinflammation in an experimental mouse stroke model. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Fine, H.A. Bevacizumab in glioblastoma--still much to learn. N. Engl. J. Med. 2014, 370, 764–765. [Google Scholar] [CrossRef]
- Immunotherapies for autoimmune diseases. Nat. Biomed. Eng. 2019, 3, 247. [CrossRef] [PubMed] [Green Version]
- Kelly, W.J.; Giles, A.J.; Gilbert, M. T lymphocyte-targeted immune checkpoint modulation in glioma. J. Immunother. Cancer 2019, 8, e000379. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Philip, M.; Rowley, D.A.; Schreiber, H. Inflammation as a tumor promoter in cancer induction. Semin. Cancer Biol. 2004, 14, 433–439. [Google Scholar] [CrossRef]
- Brenner, A.V.; Linet, M.S.; Fine, H.A.; Shapiro, W.R.; Selker, R.G.; Black, P.M.; Inskip, P.D. History of allergies and autoimmune diseases and risk of brain tumors in adults. Int. J. Cancer 2002, 99, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Anssar, T.M.; Leitzmann, M.F.; Linker, R.A.; Meier, C.; Becker, C.; Jick, S.; Sahm, K.; Platten, M.; Hau, P.; Seliger, C. Autoimmune diseases and immunosuppressive therapy in relation to the risk of glioma. Cancer Med. 2019, 9, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Cahoon, E.K.; Inskip, P.D.; Gridley, G.; Brenner, A.V. Immune-related conditions and subsequent risk of brain cancer in a cohort of 4.5 million male US veterans. Br. J. Cancer 2014, 110, 1825–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, N.; Shah, M.; Lopez-Olivo, M.; Suarez-Almazor, M.E. Use of Immune Checkpoint Inhibitors in the Treatment of Patients With Cancer and Preexisting Autoimmune Disease: A Systematic Review. Ann. Intern. Med. 2018, 168, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Pantuck, M.; McDermott, D.; Drakaki, A. To treat or not to treat: Patient exclusion in immune oncology clinical trials due to preexisting autoimmune disease. Cancer 2019, 125, 3506–3513. [Google Scholar] [CrossRef]
- Kamran, N.; Calinescu, A.; Candolfi, M.; Chandran, M.; Mineharu, Y.; Asad, A.S.; Koschmann, C.; Nunez, F.J.; Lowenstein, P.R.; Castro, M.G. Recent advances and future of immunotherapy for glioblastoma. Expert Opin. Biol. Ther. 2016, 16, 1245–1264. [Google Scholar] [CrossRef] [Green Version]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Zhu, Y.; Yao, S.; Chen, L. Cell Surface Signaling Molecules in the Control of Immune Responses: A Tide Model. Immunity 2011, 34, 466–478. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Mao, Y.; Li, M.; Lu, Y. The profile of Th17 subset in glioma. Int. Immunopharmacol. 2011, 11, 1173–1179. [Google Scholar] [CrossRef]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Murugaiyan, G.; Saha, B. Protumor vs antitumor functions of IL-17. J. Immunol. 2009, 183, 4169–4175. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol. Lett. 2005, 98, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2017, 41, 166–171. [Google Scholar] [CrossRef]
- Carvalho, J.F.; Blank, M.; Shoenfeld, Y. Vascular Endothelial Growth Factor (VEGF) in Autoimmune Diseases. J. Clin. Immunol. 2007, 27, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Dwyer, C.; Bailey, S.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Gibson, S.A.; Benveniste, E.N. Protein Kinase CK2: An Emerging Regulator of Immunity. Trends Immunol. 2018, 39, 82–85. [Google Scholar] [CrossRef]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, J. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar] [CrossRef]
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front. Immunol. 2018, 9, 2306. [Google Scholar] [CrossRef]
- Edner, N.M.; Carlesso, G.; Rush, J.S.; Walker, L.S.K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020, 19, 860–883. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Malnick, S.; Abdullah, A.; Neuman, M. Checkpoint Inhibitors and Hepatotoxicity. Biomedicines 2021, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Attarwala, H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Cencioni, M.T. The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis. Neuroimmunol. Neuroinflammation 2020, 2020, 277–290. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Janeiro, A.L.; Porciuncula, A.; Idoate-Gastearena, M.Á.; Inogés, S.; De Andrea, C.; De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Mohammadzadeh, A.; Rad, I.A.; Ahmadi-Salmasi, B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J. Neuroimmunol. 2018, 323, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Arruda, L.C.; de Azevedo, J.T.; de Oliveira, G.L.; Scortegagna, G.T.; Rodrigues, E.S.; Palma, P.V.; Brum, D.G.; Guerreiro, C.T.; Marques, V.D.; Barreira, A.; et al. Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin. Immunol. 2016, 169, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokunina-Olsson, L.; Padyukov, L.; Bennet, A.; De Faire, U.; Wiman, B.; Prince, J.; Alfredsson, L.; Klareskog, L.; Alarcón-Riquelme, M. Association of the PD-1.3A allele of thePDCD1gene in patients with rheumatoid arthritis negative for rheumatoid factor and the shared epitope. Arthritis Rheum. 2004, 50, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Hansen, D.; Husby, S.; Jacobsen, B.; Lillevang, S. Association of a putative regulatory polymorphism in the PD-1 gene with susceptibility to type 1 diabetes. Tissue Antigens 2003, 62, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Ferreirós-Vidal, I.; Gomez-Reino, J.J.; Barros, F.; Carracedo, Á.; Carreira, P.; Gonzalez-Escribano, F.; Liz, M.; Martin, J.; Ordi, J.; Vicario, J.L.; et al. Association of PDCD1 with susceptibility to systemic lupus erythematosus: Evidence of population-specific effects. Arthritis Rheum. 2004, 50, 2590–2597. [Google Scholar] [CrossRef]
- Romo-Tena, J.; Gómez-Martín, D.; Alcocer-Varela, J. CTLA-4 and autoimmunity: New insights into the dual regulator of tolerance. Autoimmun. Rev. 2013, 12, 1171–1176. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020, 20, 7. [Google Scholar] [CrossRef]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef] [PubMed]
- Dinčić, E.; Zivkovic, M.; Stanković, A.; Obradovic, D.; Alavantić, D.; Kostic, V.; Raičević, R. Association of polymorphisms in CTLA-4, IL-1ra and IL-1β genes with multiple sclerosis in Serbian population. J. Neuroimmunol. 2006, 177, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Abrams, J.R.; Kelley, S.L.; Hayes, E.; Kikuchi, T.; Brown, M.J.; Kang, S.; Lebwohl, M.G.; Guzzo, C.A.; Jegasothy, B.V.; Linsley, P.S.; et al. Blockade of T Lymphocyte Costimulation with Cytotoxic T Lymphocyte–Associated Antigen 4–Immunoglobulin (Ctla4ig) Reverses the Cellular Pathology of Psoriatic Plaques, Including the Activation of Keratinocytes, Dendritic Cells, and Endothelial Cells. J. Exp. Med. 2000, 192, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Abrams, J.R.; Lebwohl, M.G.; Guzzo, C.A.; Jegasothy, B.V.; Goldfarb, M.T.; Goffe, B.S.; Menter, A.; Lowe, N.J.; Krueger, G.; Brown, M.J.; et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J. Clin. Investig. 1999, 103, 1243–1252. [Google Scholar] [CrossRef]
- Moreland, L.W.; Alten, R.; Bosch, F.V.D.; Appelboom, T.; Leon, M.; Emery, P.; Cohen, S.; Luggen, M.; Shergy, W.J.; Nuamah, I.; et al. Costimulatory blockade in patients with rheumatoid arthritis: A pilot, dose-finding, double-blind, placebo-controlled clinical trial evaluating CTLA-4Ig and LEA29Y eighty-five days after the first infusion. Arthritis Rheum. 2002, 46, 1470–1479. [Google Scholar] [CrossRef]
- Kremer, J.M.; Dougados, M.; Emery, P.; Durez, P.; Sibilia, J.; Shergy, W.; Steinfeld, S.; Tindall, E.; Becker, J.-C.; Li, T.; et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: Twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005, 52, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Viglietta, V.; Bourcier, K.; Buckle, G.J.; Healy, B.; Weiner, H.L.; Hafler, D.A.; Egorova, S.; Guttmann, C.; Rusche, J.R.; Khoury, S. CTLA4Ig treatment in patients with multiple sclerosis: An open-label, phase 1 clinical trial. Neurology 2008, 71, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in Regulatory T Cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef] [PubMed]
- Mair, M.J.; Kiesel, B.; Feldmann, K.; Widhalm, G.; Dieckmann, K.; Wöhrer, A.; Müllauer, L.; Preusser, M.; Berghoff, A.S. LAG-3 expression in the inflammatory microenvironment of glioma. J. Neuro Oncol. 2021, 152, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Angin, M.; Brignone, C.; Triebel, F. A LAG-3–Specific Agonist Antibody for the Treatment of T Cell–Induced Autoimmune Diseases. J. Immunol. 2020, 204, 810–818. [Google Scholar] [CrossRef]
- Li, Z.; Ju, Z.; Frieri, M. The T-cell immunoglobulin and mucin domain (Tim) gene family in asthma, allergy, and autoimmunity. Allergy Asthma Proc. 2013, 34, 21–26. [Google Scholar] [CrossRef]
- Phong, B.L.; Avery, L.; Sumpter, T.L.; Gorman, J.V.; Watkins, S.; Colgan, J.; Kane, L.P. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J. Exp. Med. 2015, 212, 2289–2304. [Google Scholar] [CrossRef] [Green Version]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIMgenes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y.; et al. Molecular and clinical characterization of TIM-3 in glioma through 1024 samples. OncoImmunology 2017, 6, e1328339. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134, 570. [Google Scholar] [CrossRef]
- Chen, Y.; Langrish, C.L.; McKenzie, B.; Joyce-Shaikh, B.; Stumhofer, J.S.; McClanahan, T.; Blumenschein, W.; Churakovsa, T.; Low, J.; Presta, L.; et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Investig. 2006, 116, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambrano-Zaragoza, J.F.; Romo-Martinez, E.J.; Durán-Avelar, M.D.J.; García-Magallanes, N.; Vibanco-Pérez, N. Th17 Cells in Autoimmune and Infectious Diseases. Int. J. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Koguchi, K.; Anderson, D.E.; Yang, L.; O’Connor, K.C.; Kuchroo, V.K.; Hafler, D.A. Dysregulated T cell expression of TIM3 in multiple sclerosis. J. Exp. Med. 2006, 203, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 2009, 39, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; A Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, T.; Sato, S.; Kato, J.; Ito, Y.; Watanabe, T.; Tsuji, I.; Hori, A.; Kurokawa, T.; Kokubo, T. Nectin-2 is a potential target for antibody therapy of breast and ovarian cancers. Mol. Cancer 2013, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; DeChant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Sloan, K.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; E Roy, J.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Lucca, L.E.; Lerner, B.A.; Park, C.; DeBartolo, D.; Harnett, B.; Kumar, V.P.; Ponath, G.; Raddassi, K.; Huttner, A.; Hafler, D.A.; et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurol. Neuroimmunol. Neuroinflammation 2020, 7, e712. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.-X.; Zhang, K.; Qiu, X.-S.; Zhou, M.; Li, W.-M. CD226 Gly307Ser association with multiple autoimmune diseases: A meta-analysis. Hum. Immunol. 2013, 74, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting Edge: TIGIT Has T Cell-Intrinsic Inhibitory Functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Joller, N.; Cao, Y.; Kuchroo, V.K.; Hafler, D.A. The CD226/CD155 Interaction Regulates the Proinflammatory (Th1/Th17)/Anti-Inflammatory (Th2) Balance in Humans. J. Immunol. 2013, 191, 3673–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fife, B.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Liu, H.; Yin, G.; Xie, Q. Immunotherapy Deriving from CAR-T Cell Treatment in Autoimmune Diseases. J. Immunol. Res. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric antigen receptor T-cell therapy in glioblastoma: Charging the T cells to fight. J. Transl. Med. 2020, 18, 1–13. [Google Scholar] [CrossRef]
- Basdeo, S.A.; Cluxton, D.; Sulaimani, J.; Moran, B.; Canavan, M.; Orr, C.; Veale, D.J.; Fearon, U.; Fletcher, J.M. Ex-Th17 (Nonclassical Th1) Cells Are Functionally Distinct from Classical Th1 and Th17 Cells and Are Not Constrained by Regulatory T Cells. J. Immunol. 2017, 198, 2249–2259. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, S. The transient nature of the Th17 phenotype. Eur. J. Immunol. 2010, 40, 3312–3316. [Google Scholar] [CrossRef] [PubMed]
- Loos, J.; Schmaul, S.; Noll, T.M.; Paterka, M.; Schillner, M.; Löffel, J.T.; Zipp, F.; Bittner, S. Functional characteristics of Th1, Th17, and ex-Th17 cells in EAE revealed by intravital two-photon microscopy. J. Neuroinflammation 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parajuli, P. Role of IL-17 in Glioma Progression. J. Spine Neurosurg. 2013, 2013. [Google Scholar] [CrossRef]
- Song, Y.; Yang, J.M. Role of interleukin (IL)-17 and T-helper (Th)17 cells in cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1–8. [Google Scholar] [CrossRef]
- Paladugu, M.; Thakur, A.; Lum, L.G.; Mittal, S.; Parajuli, P. Generation and immunologic functions of Th17 cells in malignant gliomas. Cancer Immunol. Immunother. 2012, 62, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, G.; Pisati, F.; Mastropietro, A.; Frattini, V.; Iwakura, Y.; Finocchiaro, G.; Pellegatta, S. A critical role for regulatory T cells in driving cytokine profiles of Th17 cells and their modulation of glioma microenvironment. Cancer Immunol. Immunother. 2011, 60, 1739–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.; Heink, S.; Pagenstecher, A.; Reinhard, K.; Ritter, J.; Visekruna, A.; Guralnik, A.; Bollig, N.; Jeltsch, K.; Heinemann, C.; et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J. Clin. Investig. 2012, 123, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Aphale, A.; Vatan, L.; Szeliga, W.; Wang, Y.; Liu, Y.; Welling, T.H.; et al. Induction of IL-17+ T Cell Trafficking and Development by IFN-γ: Mechanism and Pathological Relevance in Psoriasis. J. Immunol. 2008, 181, 4733–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelen, E.; Thewissen, M.; Knippenberg, S.; Smolders, J.; Muris, A.-H.; Menheere, P.; Tervaert, J.C.; Hupperts, R.; Damoiseaux, J. Fraction of IL-10+ and IL-17+ CD8 T cells is increased in MS patients in remission and during a relapse, but is not influenced by immune modulators. J. Neuroimmunol. 2013, 258, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Gomes, V.; Duarte, C.; Pedreiro, S.; Carvalheiro, T.; Areias, M.; Caseiro, A.; Gabriel, A.J.; Laranjeira, P.; Pais, M.L.; et al. Distribution and functional plasticity of peripheral blood Th(c)17 and Th(c)1 in rheumatoid arthritis. Rheumatol. Int. 2013, 33, 2093–2099. [Google Scholar] [CrossRef]
- Henriques, A.; Inês, L.S.; Couto, M.; Pedreiro, S.; Santos, C.; Magalhães, M.; Santos, P.R.; Velada, I.; Almeida, A.; Carvalheiro, T.; et al. Frequency and functional activity of Th17, Tc17 and other T-cell subsets in Systemic Lupus Erythematosus. Cell. Immunol. 2010, 264, 97–103. [Google Scholar] [CrossRef]
- Li, J.; Huang, Z.-F.; Xiong, G.; Mo, H.-Y.; Qiu, F.; Mai, H.-Q.; Chen, Q.-Y.; He, J.; Chen, S.-P.; Zheng, L.-M.; et al. Distribution, characterization, and induction of CD8+ regulatory T cells and IL-17-producing CD8+ T cells in nasopharyngeal carcinoma. J. Transl. Med. 2011, 9, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Hernandez, M.D.L.L.; Hamada, H.; Reome, J.B.; Misra, S.K.; Tighe, M.P.; Dutton, R.W. Adoptive Transfer of Tumor-Specific Tc17 Effector T Cells Controls the Growth of B16 Melanoma in Mice. J. Immunol. 2010, 184, 4215–4227. [Google Scholar] [CrossRef] [Green Version]
- Sestero, C.M.; McGuire, D.; De Sarno, P.; Brantley, E.C.; Soldevila, G.; Axtell, R.C.; Raman, C. CD5-dependent CK2 activation pathway regulates threshold for T cell anergy. J. Immunol. 2012, 189, 2918–2930. [Google Scholar] [CrossRef] [Green Version]
- Axtell, R.C.; Xu, L.; Barnum, S.R.; Raman, C. CD5-CK2 Binding/Activation-Deficient Mice Are Resistant to Experimental Autoimmune Encephalomyelitis: Protection Is Associated with Diminished Populations of IL-17-Expressing T Cells in the Central Nervous System. J. Immunol. 2006, 177, 8542–8549. [Google Scholar] [CrossRef] [Green Version]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Bühler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef] [Green Version]
- Dubois, N.; Willems, M.; Nguyen-Khac, M.-T.; Kroonen, J.; Goffart, N.; Deprez, M.; Bours, V.; Robe, P.A. Constitutive activation of casein kinase 2 in glioblastomas: Absence of class restriction and broad therapeutic potential. Int. J. Oncol. 2016, 48, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Di Maggio, S.A.; Cabrelle, A.; et al. Protein Kinase CK2 Inhibition Down Modulates the NF-κB and STAT3 Survival Pathways, Enhances the Cellular Proteotoxic Stress and Synergistically Boosts the Cytotoxic Effect of Bortezomib on Multiple Myeloma and Mantle Cell Lymphoma Cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor Necrosis Factor α-induced Phosphorylation of RelA/p65 on Ser529 Is Controlled by Casein Kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, B.; Fischer, M.; Schaefer, S.; Issinger, O.-G. The kinase inhibitor D11 induces caspase-mediated cell death in cancer cells resistant to chemotherapeutic treatment. J. Exp. Clin. Cancer Res. 2015, 34, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgo, C.; Ruzzene, M. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mirshafiey, A.; Mohsenzadegan, M. TGF-β as a promising option in the treatment of multiple sclerosis. Neuropharmacology 2009, 56, 929–936. [Google Scholar] [CrossRef]
- YiKim, I.; Kim, M.M.; Kim, S.-J. Transforming Growth Factor-β: Biology and Clinical Relevance. BMB Rep. 2005, 38, 1–8. [Google Scholar] [CrossRef]
- Ihara, S.; Hirata, Y.; Koike, K. TGF-β in inflammatory bowel disease: A key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 2017, 52, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; A Alvarez-Breckenridge, C.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFβ2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers 2019, 11, 1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-β signaling pathway in human cancer. Expert Opin. Investig. Drugs 2009, 19, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Ishigame, H.; Zenewicz, L.A.; Sanjabi, S.; Licona-Limón, P.; Nakayama, M.; Leonard, W.J.; Flavell, R.A. Excessive Th1 responses due to the absence of TGF- signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, 6961–6966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabresi, P.A.; Fields, N.S.; Maloni, H.W.; Hanham, A.; Carlino, J.; Moore, J.; Levin, M.; Dhib-Jalbut, S.; Tranquill, L.R.; Austin, H.; et al. Phase 1 trial of transforming growth factor beta 2 in chronic progressive MS. Neurology 1998, 51, 289–292. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an Oral SMAD7 Antisense Oligonucleotide, and Crohn’s Disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C. The microbiota-immune axis as a central mediator of gut-brain communication. Neurobiol. Dis. 2020, 136, 104714. [Google Scholar] [CrossRef]
- Lee, P.W.; Severin, M.E.; Lovett-Racke, A.E. TGF-β regulation of encephalitogenic and regulatory T cells in multiple sclerosis. Eur. J. Immunol. 2017, 47, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.-P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Rossin, A.; Miloro, G.; Hueber, A.-O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Jiang, Z.; Li, X.; Xu, Y.; Shao, Z. Cytokines: Shifting the balance between glioma cells and tumor microenvironment after irradiation. J. Cancer Res. Clin. Oncol. 2014, 141, 575–589. [Google Scholar] [CrossRef]
- Hao, C.; Beguinot, F.; Condorelli, G.; Trencia, A.; Van Meir, E.G.; Yong, V.W.; Parney, I.; Roa, W.H.; Petruk, K.C. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001, 61, 1162–1170. [Google Scholar]
- Nitsch, R.; Bechmann, I.; A Deisz, R.; Haas, D.; Lehmann, T.N.; Wendling, U.; Zipp, F. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 2000, 356, 827–828. [Google Scholar] [CrossRef]
- Aktas, O. The role of TRAIL/TRAIL receptors in central nervous system pathology. Front. Biosci. 2007, 12, 2912–2921. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Stagg, J.; Yagita, H.; Okumura, K.; Smyth, M. Targeting death-inducing receptors in cancer therapy. Oncogene 2007, 26, 3745–3757. [Google Scholar] [CrossRef] [Green Version]
- Yuan, K.; Sun, Y.; Zhou, T.; McDonald, J.M.; Chen, Y. PARP-1 Regulates Resistance of Pancreatic Cancer to TRAIL Therapy. Clin. Cancer Res. 2013, 19, 4750–4759. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, P.; LeQuesne, J.; Grellard, J.-M.; Dugué, A.; Coquan, E.; Brachet, P.-E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Cretney, E.; McQualter, J.L.; Kayagaki, N.; Yagita, H.; Bernard, C.C.A.; Grewal, I.; Ashkenazi, A.; Smyth, M.J. TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L suppresses experimental autoimmune encephalomyelitis in mice. Immunol. Cell Biol. 2005, 83, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Hirata, S.; Fukushima, S.; Matsunaga, Y.; Ito, T.; Uchino, M.; Nishimura, Y.; Senju, S. Dual Effects of TRAIL in Suppression of Autoimmunity: The Inhibition of Th1 Cells and the Promotion of Regulatory T Cells. J. Immunol. 2010, 185, 5259–5267. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.-E.; Zheng, S.-J.; Maguschak, K.A.; Peschon, J.; Chen, Y.H. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol. 2003, 4, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Chen, Y.; Göke, R.; Wilmen, A.; Seidel, C.; Göke, A.; Hilliard, B.; Chen, Y. Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (Trail) Is an Inhibitor of Autoimmune Inflammation and Cell Cycle Progression. J. Exp. Med. 2000, 191, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Aktas, O.; Smorodchenko, A.; Brocke, S.; Infante-Duarte, C.; Topphoff, U.S.; Vogt, J.; Prozorovski, T.; Meier, S.; Osmanova, V.; Pohl, E.; et al. Neuronal Damage in Autoimmune Neuroinflammation Mediated by the Death Ligand TRAIL. Neuron 2005, 46, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendling, U.; Walczak, H.; Dörr, J.; Jaboci, C.; Weller, M.; Krammer, P.H.; Zipp, F. Expression of TRAIL receptors in human autoreactive and foreign antigen-specific T cells. Cell Death Differ. 2000, 7, 637–644. [Google Scholar] [CrossRef]
- Plate, K.H.; Warnke, P.C. Vascular endothelial growth factor. J. Neuro-Oncology 1997, 35, 363–370. [Google Scholar] [CrossRef]
- Garcia-Romero, N.; Aliana, I.P.; Madurga, R.; Carrión-Navarro, J.; Esteban-Rubio, S.; Jiménez, B.; Collazo, A.; Pérez-Rodríguez, F.; De Mendivil, A.O.; Fernández-Carballal, C.; et al. Bevacizumab dose adjustment to improve clinical outcomes of glioblastoma. BMC Med. 2020, 18, 1–16. [Google Scholar] [CrossRef]
- Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J. Clin. Med. 2020, 9, 675. [Google Scholar] [CrossRef]
- Jain, R.K.; Di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef]
- Stanković, N.D.; Bicker, F.; Keller, S.; Jones, D.T.W.; Harter, P.N.; Kienzle, A.; Gillmann, C.; Arnold, P.; Golebiewska, A.; Keunen, O.; et al. EGFL7 enhances surface expression of integrin α 5 β 1 to promote angiogenesis in malignant brain tumors. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Tamura, R.; Morimoto, Y.; Kosugi, K.; Sato, M.; Oishi, Y.; Ueda, R.; Kikuchi, R.; Nagashima, H.; Hikichi, T.; Noji, S.; et al. Clinical and histopathological analyses of VEGF receptors peptide vaccine in patients with primary glioblastoma—A case series. BMC Cancer 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zuo, W.; Yang, P.; Zhang, Y. Anti-PD-1, anti-VEGF, and temozolomide therapy in a patient with recurrent glioblastoma: A case report. J. Int. Med Res. 2020, 48, 0300060520951395. [Google Scholar] [CrossRef]
- Mealy, M.A.; Shin, K.; John, G.; Levy, M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin. Exp. Neuroimmunol. 2015, 6, 413–418. [Google Scholar] [CrossRef]
- Casan, J.M.L.; Wong, J.; Northcott, M.J.; Opat, S. Anti-CD20 monoclonal antibodies: Reviewing a revolution. Hum. Vaccines Immunother. 2018, 14, 2820–2841. [Google Scholar] [CrossRef]
- Wong, E.T.; Tishler, R.; Barron, L.; Wu, J.K. Immunochemotherapy with rituximab and temozolomide for central nervous system lymphomas. Cancer 2004, 101, 139–145. [Google Scholar] [CrossRef]
- Boye, J.; Elter, T.; Engert, A. An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann. Oncol. 2003, 14, 520–535. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Rashidi, A.; Miska, J.; Zhang, P.; Pituch, K.C.; Hou, D.; Xiao, T.; Fischietti, M.; Kang, S.J.; Appin, C.L.; et al. Myeloid-Derived Suppressive Cells Promote B cell–Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol. Res. 2019, 7, 1928–1943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Wang, H.; Song, S.W. Identification of a five B cell-associated gene prognostic and predictive signature for advanced glioma patients harboring immunosuppressive subtype preference. Oncotarget 2016, 7, 73971–73983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abboud, H.; Probasco, J.C.; Irani, S.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune encephalitis: Proposed best practice recommendations for diagnosis and acute management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 757–768. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of Neuromyelitis Optica With Rituximab: Retrospective Analysis of 25 Patients. Arch. Neurol. 2008, 65, 1443–1448. [Google Scholar] [CrossRef]
- Nowak, R.J.; DiCapua, D.B.; Zebardast, N.; Goldstein, J.M. Response of patients with refractory myasthenia gravis to rituximab: A retrospective study. Ther. Adv. Neurol. Disord. 2011, 4, 259–266. [Google Scholar] [CrossRef]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. J. Neuro Oncol. 2021, 151, 375–391. [Google Scholar] [CrossRef]
- De Felice, F.; Pranno, N.; Marampon, F.; Musio, D.; Salducci, M.; Polimeni, A.; Tombolini, V. Immune check-point in glioblastoma multiforme. Crit. Rev. Oncol. 2019, 138, 60–69. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Abbott, R.C.; Verdon, D.J.; Gracey, F.M.; E Hughes-Parry, H.; Iliopoulos, M.; A Watson, K.; Mulazzani, M.; Luong, K.; D’Arcy, C.; Sullivan, L.C.; et al. Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin. Transl. Immunol. 2021, 10, e1283. [Google Scholar] [CrossRef]
- Stylli, S.S. Novel Treatment Strategies for Glioblastoma. Cancers 2020, 12, 2883. [Google Scholar] [CrossRef] [PubMed]
- Lynes, J.P.; Nwankwo, A.K.; Sur, H.P.; E Sanchez, V.; Sarpong, K.A.; Ariyo, O.I.; Dominah, G.A.; Nduom, E.K. Biomarkers for immunotherapy for treatment of glioblastoma. J. Immunother. Cancer 2019, 8, e000348. [Google Scholar] [CrossRef]
- Wang, J.; Shen, F.; Yao, Y.; Wang, L.-L.; Zhu, Y.; Hu, J. Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma. Front. Oncol. 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Zheng, Y.; Hong, W.; Chen, X.; Li, H.; Huang, B.; Huang, Z.; Tang, H.; Geng, W. Recent Advances in Immune Cell Therapy for Glioblastoma. Front. Immunol. 2020, 11, 544563. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Chekhonin, V.P. Recent Advances in Oncolytic Virotherapy and Immunotherapy for Glioblastoma: A Glimmer of Hope in the Search for an Effective Therapy? Cancers 2018, 10, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett. 2017, 403, 224–230. [Google Scholar] [CrossRef]
- Pituch, K.C.; Zannikou, M.; Ilut, L.; Xiao, T.; Chastkofsky, M.; Sukhanova, M.; Bertolino, N.; Procissi, D.; Amidei, C.; Horbinski, C.M.; et al. Neural stem cells secreting bispecific T cell engager to induce selective antiglioma activity. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Valencia, J.C.; Egbukichi, N.; Erwin-Cohen, R.A. Autoimmunity and Cancer, the Paradox Comorbidities Challenging Therapy in the Context of Preexisting Autoimmunity. J. Interf. Cytokine Res. 2019, 39, 72–84. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; E LaCouture, M.; A Postow, M.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 1–20. [Google Scholar] [CrossRef]
- Jia, X.-H.; Geng, L.-Y.; Jiang, P.-P.; Xu, H.; Nan, K.-J.; Yao, Y.; Jiang, L.-L.; Sun, H.; Qin, T.-J.; Guo, H. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J. Exp. Clin. Cancer Res. 2020, 39, 284. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Eun, Y.; Kim, I.Y.; Sun, J.-M.; Lee, J.; Cha, H.-S.; Koh, E.-M.; Kim, H.; Lee, J. Risk factors for immune-related adverse events associated with anti-PD-1 pembrolizumab. Sci. Rep. 2019, 9, 14039. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; A Mitchell, D.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.H.; Woroniecka, K.; Barbour, A.B.; Fecci, P.E.; Sanchez-Perez, L.; Sampson, J.H. CAR T cells and checkpoint inhibition for the treatment of glioblastoma. Expert Opin. Biol. Ther. 2020, 20, 579–591. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Targeted Molecule | G | AD | Phase | Treatment | Study Number |
|---|---|---|---|---|---|
| PD-1 | X | III | E: Nivolumab + TMZ + RT C: Nivolumab Placebo + TMZ + RT | NCT02667587 | |
| X | II | Neoadjuvant Nivolumab | NCT02550249 | ||
| X | II | Prior in all groups: RT + TMZ E: Nivolumab + TMZ Control: TMZ alone | NCT04195139 | ||
| CTLA-4 | X | II/III | E: Nivolumab + Ipilimumab + RT C: TMZ + RT | NCT04396860 | |
| X | II | E: Abatacept followed by placebo C: placebo followed by Abatacept | NCT01116427 | ||
| X | II | E: Ipilimumab + Nivolumab followed by Nivolumab alone | NCT04145115 | ||
| X | I/II | E1: CTLA4-Ig + Cyclophosphamide E2: CTLA4-Ig + Cyclophosphamide Control: Cyclophosphamide alone | NCT00094380 | ||
| X | I | E1: Nivolumab + placebo followed by Nivolumab alone E2: Nivolumab + Ipilimumab followed by Nivolumab alone E3: Placebo + Ipilimumab followed by Nivolumab alone | NCT04323046 | ||
| X | I | E: CTLA4-Ig | NCT00076934 | ||
| X | I | E1: Nivolumab + Ipilimumab followed by IL13Ralpha2-CAR T cells + Nivolumab E2: IL13Ralpha2-CAR T cells + Nivolumab | NCT04003649 | ||
| X | I/II | E: Belatacept/Abatacept (multiple doses) | NCT00279760 | ||
| X | I | E: Ipilimumab (intra-tumoral) + Nivolumab (intravenous) | NCT03233152 | ||
| LAG-3 | X | I | E A1: anti-LAG-3 E A2: Urelumab E B1: anti-LAG-3 + Nivolumab E B2: Nivolumab + Urelumab E I: patients receive pre-operatively and 45 days after surgery a drug from one of the four arms mentioned above | NCT02658981 | |
| X | I | E: anti-LAG-3 + Nivolumab | NCT03493932 | ||
| X | Prep | anti-LAG-3 (patent no. 3344654) | |||
| CK2 | X | I/II | CK-2 inhibitor in recurrent medulloblastoma E I: children E II: adults E S: before surgery in subjects from I and II | NCT03904862 | |
| TIGIT | X | 0/I | E A: anti-TIGIT + anti-PD-1 (Safety Cohort) E B1: anti-TIGIT + placebo (Surgical Cohort) E B2: anti-PD-1 + placebo (Surgical Cohort) E B3: anti-TIGIT + anti-PD-1 (Surgical Cohort) E B4: placebo (Surgical Cohort) all Experimental B followed by anti-TIGIT + anti-PD-1 | NCT04656535 | |
| TIM-3 | X | I | E: anti-TIM-3 + anti-PD-1 + radiation therapy | NCT03961971 | |
| TGF-β | X | Ib/IIa | E IA: RT + 80 mg TGF-β inhibitor + TMZ followed by TGF-β inhibitor + TMZ E IB: RT + 150 mg TGF-β inhibitor + TMZ followed by TGF-β inhibitor + TMZ E II: RT + established dose from I of TGF-β inhibitor + TMZ followed by TGF-β inhibitor + TMZ Control: RT + TMZ followed by TMZ alone | NCT01220271 | |
| TRAIL | X | I/IIa | E: Olaparib + TMZ + RT followed by Olaparib alone, then Olaparib + TMZ | NCT03212742 | |
| VEGF | X | II | E 1: saline + Aflibercept E 2: hyaluronidase + Aflibercept E 3: hyaluronidase alone | NCT04311606 | |
| X | II | C 1: Bevacizumab + radiation (naive recurrent grade IV gliomas) C 2: Bevacizumab + radiation (exposed and refractive grade IV gliomas) C 3: Bevacizumab + radiation (naive recurrent grade III gliomas) C 4: Bevacizumab + radiation (exposed and refractive grade III gliomas) | NCT01743950 | ||
| X | I/II | E: Cetuximab + Bevacizumab via Superselective Intraarterial Cerebral Infusion | NCT01884740 | ||
| X | I/II | E: peptide vaccine of VEGFR (subcutaneous) | UMIN000013381 | ||
| CD20 | X | IV | E: Rituximab (standard infusion + rapid infusion) | NCT02040116 | |
| X | III | C 1: Rituximab (infusion) C 2: Cladribine (oral) | NCT04121403 | ||
| X | II | E: Rituximab followed by Rituximab C: Placebo followed by Rituximab | NCT04274257 | ||
| X | II | E 1: Rituximab (intrathecal) + methylprednisolone (intravenous) E 2: Rituximab (intrathecal) + methylprednisolone (intravenous) + Rituximab (intravenous) C: methylprednisolone (intravenous) | NCT02545959 | ||
| X | II | E: Rituximab (intravenous) C: placebo (intravenous) | NCT00279305 | ||
| X | I/II | E: Rituximab (intravenous) | NCT00036491 | ||
| X | I | E: Rituximab (2 times intravenous) | NCT01086631 | ||
| X | I | E: Rituximab (2 times intravenous) | NCT00101829 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitar, L.; Schumann, U.; König, R.; Zipp, F.; Schmidt, M.H.H. Targeting Immune Modulators in Glioma While Avoiding Autoimmune Conditions. Cancers 2021, 13, 3524. https://doi.org/10.3390/cancers13143524
Bitar L, Schumann U, König R, Zipp F, Schmidt MHH. Targeting Immune Modulators in Glioma While Avoiding Autoimmune Conditions. Cancers. 2021; 13(14):3524. https://doi.org/10.3390/cancers13143524
Chicago/Turabian StyleBitar, Lynn, Ulrike Schumann, Renate König, Frauke Zipp, and Mirko H. H. Schmidt. 2021. "Targeting Immune Modulators in Glioma While Avoiding Autoimmune Conditions" Cancers 13, no. 14: 3524. https://doi.org/10.3390/cancers13143524
APA StyleBitar, L., Schumann, U., König, R., Zipp, F., & Schmidt, M. H. H. (2021). Targeting Immune Modulators in Glioma While Avoiding Autoimmune Conditions. Cancers, 13(14), 3524. https://doi.org/10.3390/cancers13143524