
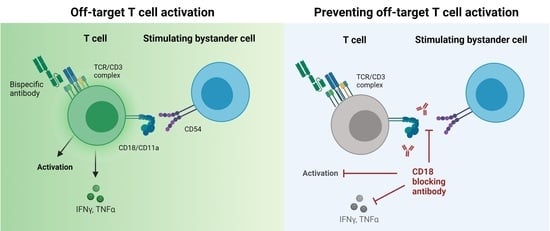
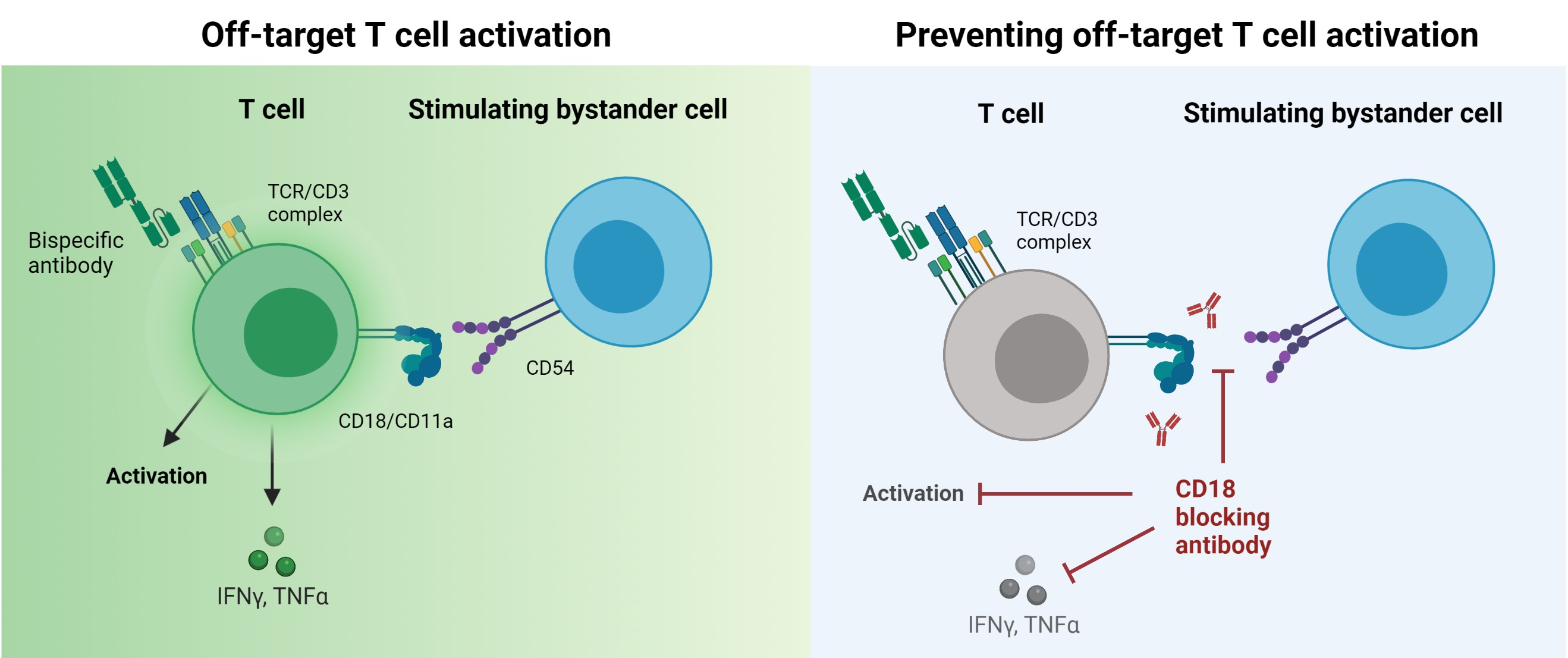
CD18 Antibody Application Blocks Unwanted Off-Target T Cell Activation Caused by Bispecific Antibodies

 , , ,
, , ,  ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Antibodies and Flow Cytometry
2.3. Real-Time Tumor Cell Killing Assay
2.4. Legendplex Cytokine Arrays
2.5. Statistical Analysis
3. Results
3.1. Off-Target T Cell Activation upon bsAb Binding in the Absence of Target Cells
3.2. Endothelial and Lymphoid Cells Enhance Off-Target T Cell Activation by Acting as Stimulating Bystanders
3.3. Integrin Ligands Are Involved in BsAb-mediated Off-Target T Cell Activation
3.4. BsAb-mediated Off-Target T Cell Activation Is Prevented by Integrin Blockade
3.5. Blockade of CD2 and CD18 Differently Affects On-Target T Cell Activation
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef]
- Jung, G.; Honsik, C.J.; Reisfeld, R.A.; Müller-Eberhard, H.J. Activation of human peripheral blood mononuclear cells by anti-T3: Killing of tumor target cells coated with anti-target-anti-T3 conjugates. Proc. Natl. Acad. Sci. USA 1986, 83, 4479–4483. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.; Eberhard, H.J.M. An in-vitro model for tumor immunotherapy with antibody heteroconjugates. Immunol. Today 1988, 9, 257–260. [Google Scholar] [CrossRef]
- Jung, G.; Freimann, U.; Marschall, Z.V.; Reisfeld, R.A.; Wilmanns, W. Target cell-induced T cell activation with bi-and trispecific antibody fragments. Eur. J. Immunol. 1991, 21, 2431–2435. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.-A.; Raff, T.; Viardot, A.; Schmid, M. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, B.; Buter, J.; Sleijfer, D.T.; Janssen, R.; Van der Graaf, W.; The, T.; De Leij, L.; Mulder, N. Phase I study of intravenously applied bispecific antibody in renal cell cancer patients receiving subcutaneous interleukin 2. Br. J. Cancer 1994, 70, 652–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibben, J.G.; Boerman, O.C.; Massuger, L.F.; Schijf, C.P.; Claessens, R.A.; Corstens, F.H. Pharmacokinetics, biodistribution and biological effects of intravenously administered bispecific monoclonal antibody OC/TR F (ab′) 2 in ovarian carcinoma patients. Int. J. Cancer 1996, 66, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nübling, T.; Pyz, E.; Bühring, H.-J.; Rammensee, H.-G.; Salih, H.R.; Große-Hovest, L.; et al. Characterization of a Bispecific FLT3 X CD3 Antibody in an Improved, Recombinant Format for the Treatment of Leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zekri, L.; Vogt, F.; Osburg, L.; Müller, S.; Kauer, J.; Manz, T.; Pflügler, M.; Maurer, A.; Heitmann, J.S.; Hagelstein, I. An IgG-based bispecific antibody for improved dual targeting in PSMA-positive cancer. EMBO Mol. Med. 2021, 13, e11902. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Fenton, M.; Whiteside, T.L.; Ferrone, S.; Boyiadzis, M. Chondroitin sulfate proteoglycan-4 (CSPG4)-specific monoclonal antibody 225.28 in detection of acute myeloid leukemia blasts. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2015, 22, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell. Mol. Biol. Lett. 2017, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bannerji, R.; Arnason, J.E.; Advani, R.; Brown, J.R.; Allan, J.N.; Ansell, S.M.; Barnes, J.; O’Brien, S.M.; Chavez, J.C.; Duell, J.; et al. Emerging Clinical Activity of REGN1979, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Follicular Lymphoma (FL), Diffuse Large B-Cell Lymphoma (DLBCL), and Other B-Cell Non-Hodgkin Lymphoma (B-NHL) Subtypes. Blood 2018, 132, 1690. [Google Scholar] [CrossRef]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 dose escalation study of MEDI-565, a bispecific T-cell engager that targets human carcinoembryonic antigen, in patients with advanced gastrointestinal adenocarcinomas. Clin. Color. Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef]
- de Fougerolles, A.R.; Stacker, S.A.; Schwarting, R.; Springer, T.A. Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J. Exp. Med. 1991, 174, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Krummel, M.F.; Bartumeus, F.; Gérard, A. T cell migration, search strategies and mechanisms. Nat. Rev. Immunol. 2016, 16, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Lisowska-Grospierre, B.; Anderson, D.; Springer, T. Leukocyte adhesion deficiency: Molecular basis and functional consequences. Immunodefic. Rev. 1988, 1, 39–54. [Google Scholar]
- Walling, B.L.; Kim, M. LFA-1 in T cell migration and differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [Green Version]
- Molema, G.; Tervaert, J.C.; Kroesen, B.; Helfrich, W.; Meijer, D.; de Leij, L. CD3 directed bispecific antibodies induce increased lymphocyte–endothelial cell interactions in vitro. Br. J. Cancer 2000, 82, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Hofmann, T.J.; Gershenson, Z.; Levine, B.L.; Grupp, S.A.; Teachey, D.T.; Barrett, D.M. Monocyte lineage–derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 2017, 19, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; Van Der Stegen, S.J.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.-J.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11, eaax8861. [Google Scholar]
- Kragstrup, T.W.; Juul-Madsen, K.; Christiansen, S.H.; Zhang, X.; Krog, J.; Vorup-Jensen, T.; Kjærgaard, A.G. Altered levels of soluble CD18 may associate immune mechanisms with outcome in sepsis. Clin. Exp. Immunol. 2017, 190, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Salas, A.; Shimaoka, M.; Chen, S.; Carman, C.V.; Springer, T. Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 2002, 277, 50255–50262. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L.; Springer, T.A. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 1989, 341, 619–624. [Google Scholar] [CrossRef]
- Van Kooyk, Y.; Van de Wiel-van Kemenade, P.; Weder, P.; Kuijpers, T.; Figdor, C. Enhancement of LFA-1-mediated cell adhesion by triggering through CD2 or CD3 on T lymphocytes. Nature 1989, 342, 811–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, G.L.; Lee, S.-H.; Schubbert, S.; Miranda, Y.; Rashid, R.; Pong, E.; Phung, S.; Chan, E.W.; Chen, H.; Endo, N.; et al. Tuning T cell affinity improves efficacy and safety of anti-CD38× anti-CD3 bispecific antibodies in monkeys-a potential therapy for multiple myeloma. Blood 2015, 136, 1798. [Google Scholar] [CrossRef]
- Bonvini, E.; La Motte-Mohs, R.; Huang, L.; Lam, C.-Y.K.; Kaufman, T.; Liu, L.; Alderson, R.F.; Stahl, K.; Brown, J.G.; Li, H.; et al. A Next-Generation Fc-Bearing CD3-Engaging Bispecific DART® Platform with Extended Pharmacokinetic and Expanded Pharmacologic Window: Characterization As CD123 x CD3 and CD19 x CD3 DART Molecules. Blood 2018, 132, 5230. [Google Scholar] [CrossRef]
- Ulbrich, H.; Eriksson, E.E.; Lindbom, L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol. Sci. 2003, 24, 640–647. [Google Scholar] [CrossRef]
- Faxon, D.P.; Gibbons, R.J.; Chronos, N.A.; Gurbel, P.A.; Sheehan, F.; Investigators, H.-M. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: The results of the HALT-MI study. J. Am. Coll. Cardiol. 2002, 40, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Baran, K.W.; Nguyen, M.; McKendall, G.R.; Lambrew, C.T.; Dykstra, G.; Palmeri, S.T.; Gibbons, R.J.; Borzak, S.; Sobel, B.E.; Gourlay, S.G.; et al. Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: Limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 2001, 104, 2778–2783. [Google Scholar]
- Rusnak, J.M.; Kopecky, S.L.; Clements, I.P.; Gibbons, R.J.; Holland, A.E.; Peterman, H.S.; Martin, J.S.; Saoud, J.B.; Feldman, R.L.; Breisblatt, W.M.; et al. An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am. J. Cardiol. 2001, 88, 482–487. [Google Scholar] [CrossRef]
- Rhee, P.; Morris, J.; Durham, R.; Hauser, C.; Cipolle, M.; Wilson, R.; Luchette, F.; McSwain, N.; Miller, R. Recombinant humanized monoclonal antibody against CD18 (rhu MAb CD18) in traumatic hemorrhagic shock: Results of a phase II clinical trial. J. Trauma 2000, 49, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.D.; Petersdorf, S.H.; Richards, T.L.; Maravilla, K.R.; Dale, D.C.; Price, T.H.; John, T.P.S.; Yu, A.S. Phase I study of a humanized anti-CD11/CD18 monoclonal antibody in multiple sclerosis. Clin. Pharmacol. Ther. 1998, 64, 339–346. [Google Scholar] [CrossRef]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. New Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Yousry, T.A.; Major, E.O.; Ryschkewitsch, C.; Fahle, G.; Fischer, S.; Hou, J.; Curfman, B.; Miszkiel, K.; Mueller-Lenke, N.; Sanchez, E.; et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. New Engl. J. Med. 2006, 354, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kauer, J.; Vogt, F.; Hagelstein, I.; Hörner, S.; Märklin, M.; Maurer, S.; Salih, H.R.; Jung, G.; Zekri, L. CD18 Antibody Application Blocks Unwanted Off-Target T Cell Activation Caused by Bispecific Antibodies. Cancers 2021, 13, 4596. https://doi.org/10.3390/cancers13184596
Kauer J, Vogt F, Hagelstein I, Hörner S, Märklin M, Maurer S, Salih HR, Jung G, Zekri L. CD18 Antibody Application Blocks Unwanted Off-Target T Cell Activation Caused by Bispecific Antibodies. Cancers. 2021; 13(18):4596. https://doi.org/10.3390/cancers13184596
Chicago/Turabian StyleKauer, Joseph, Fabian Vogt, Ilona Hagelstein, Sebastian Hörner, Melanie Märklin, Stefanie Maurer, Helmut R. Salih, Gundram Jung, and Latifa Zekri. 2021. "CD18 Antibody Application Blocks Unwanted Off-Target T Cell Activation Caused by Bispecific Antibodies" Cancers 13, no. 18: 4596. https://doi.org/10.3390/cancers13184596
APA StyleKauer, J., Vogt, F., Hagelstein, I., Hörner, S., Märklin, M., Maurer, S., Salih, H. R., Jung, G., & Zekri, L. (2021). CD18 Antibody Application Blocks Unwanted Off-Target T Cell Activation Caused by Bispecific Antibodies. Cancers, 13(18), 4596. https://doi.org/10.3390/cancers13184596