
The Emerging Importance of Tumor Genomics in Operable Non-Small Cell Lung Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
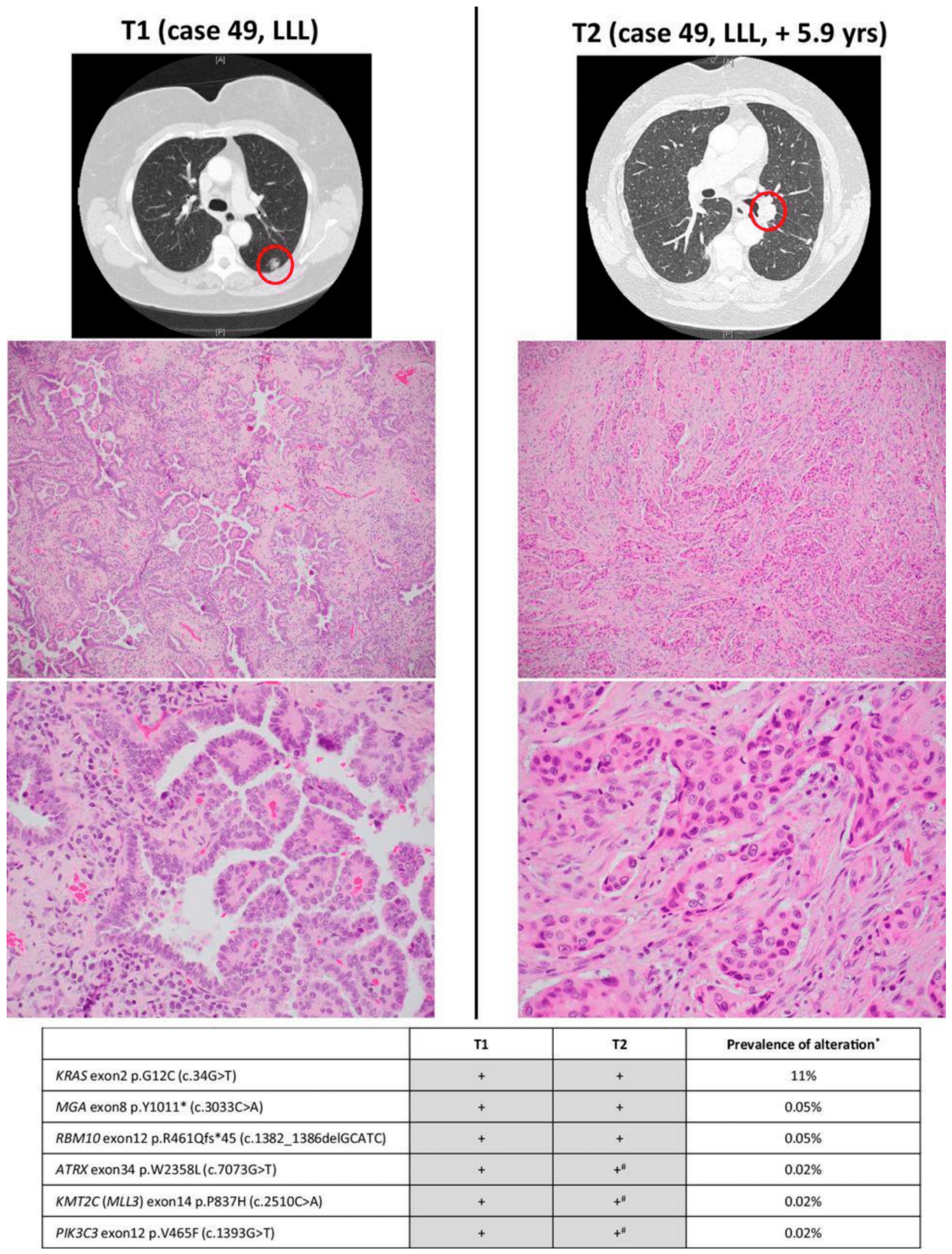
2. Tumor Heterogeneity and Evolution
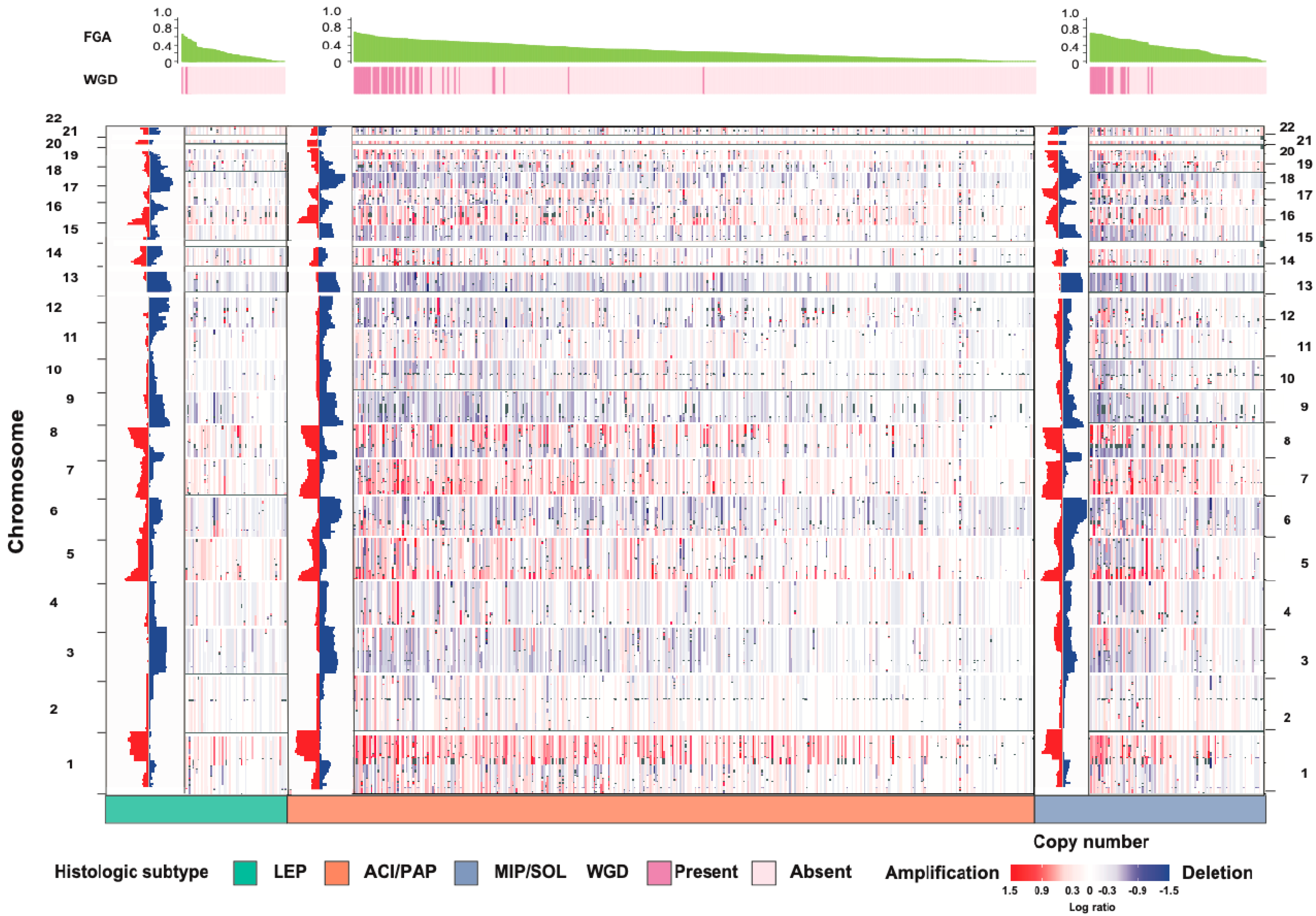
3. Understanding the Biology of LUAD Histologic Subtypes
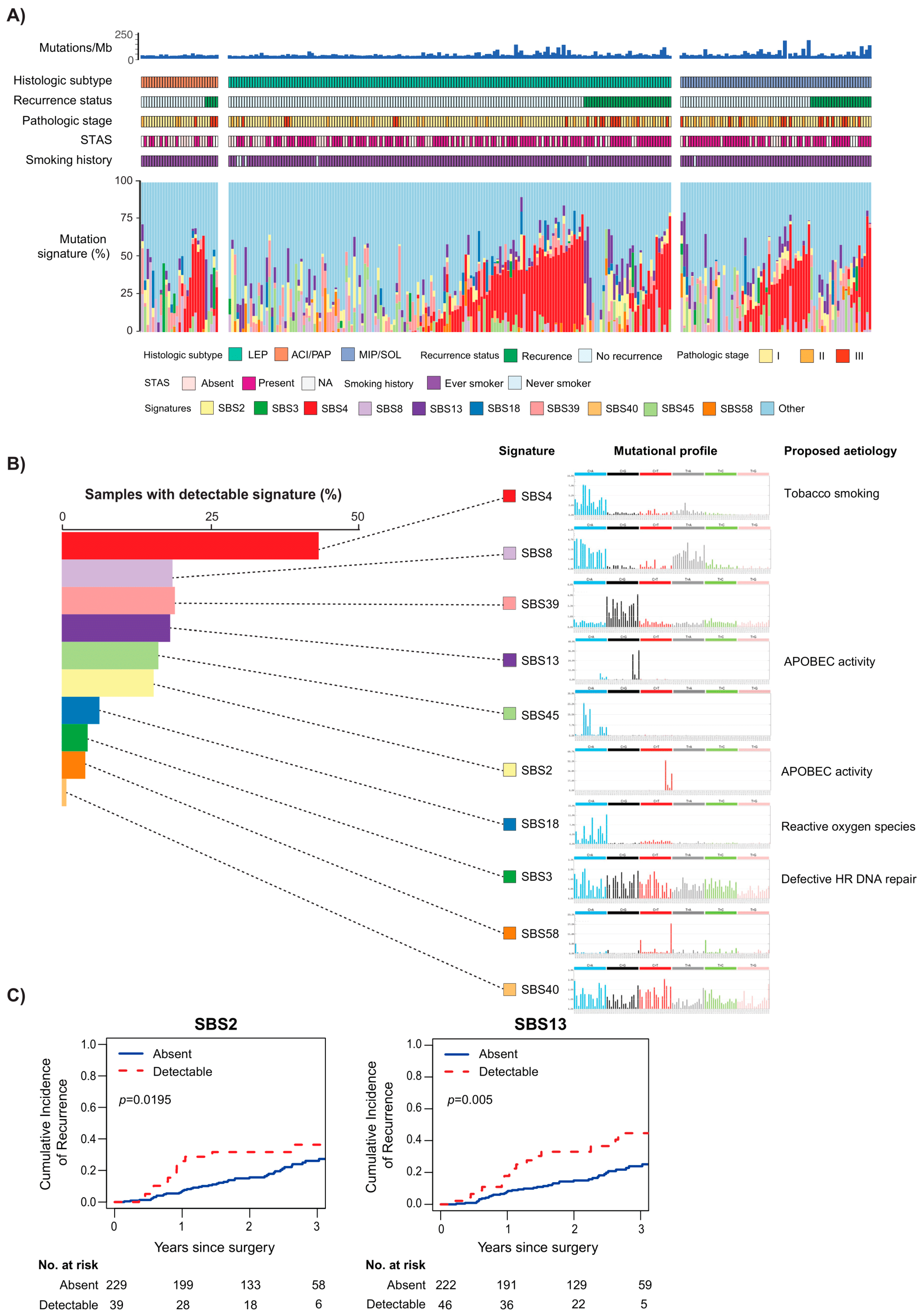
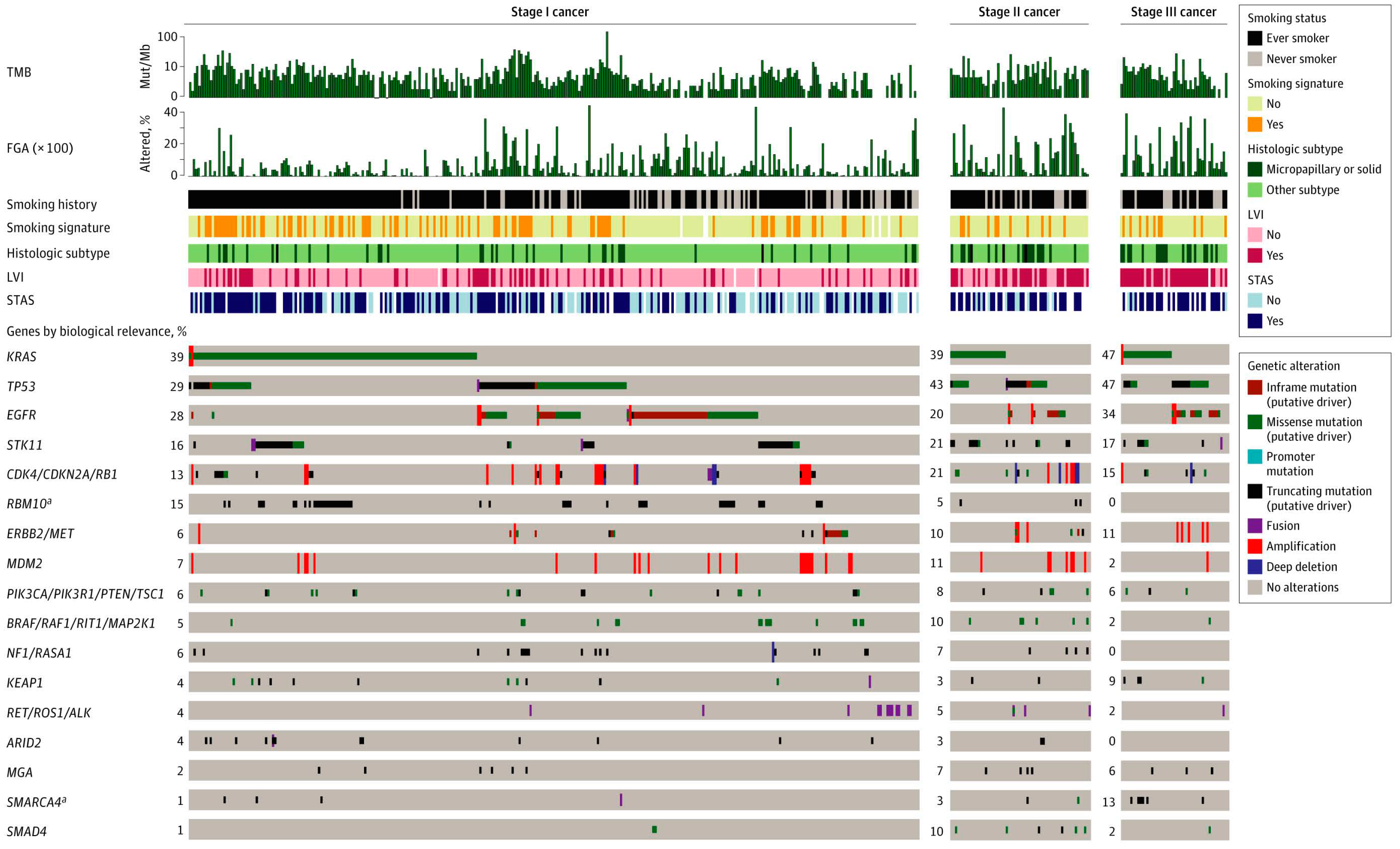
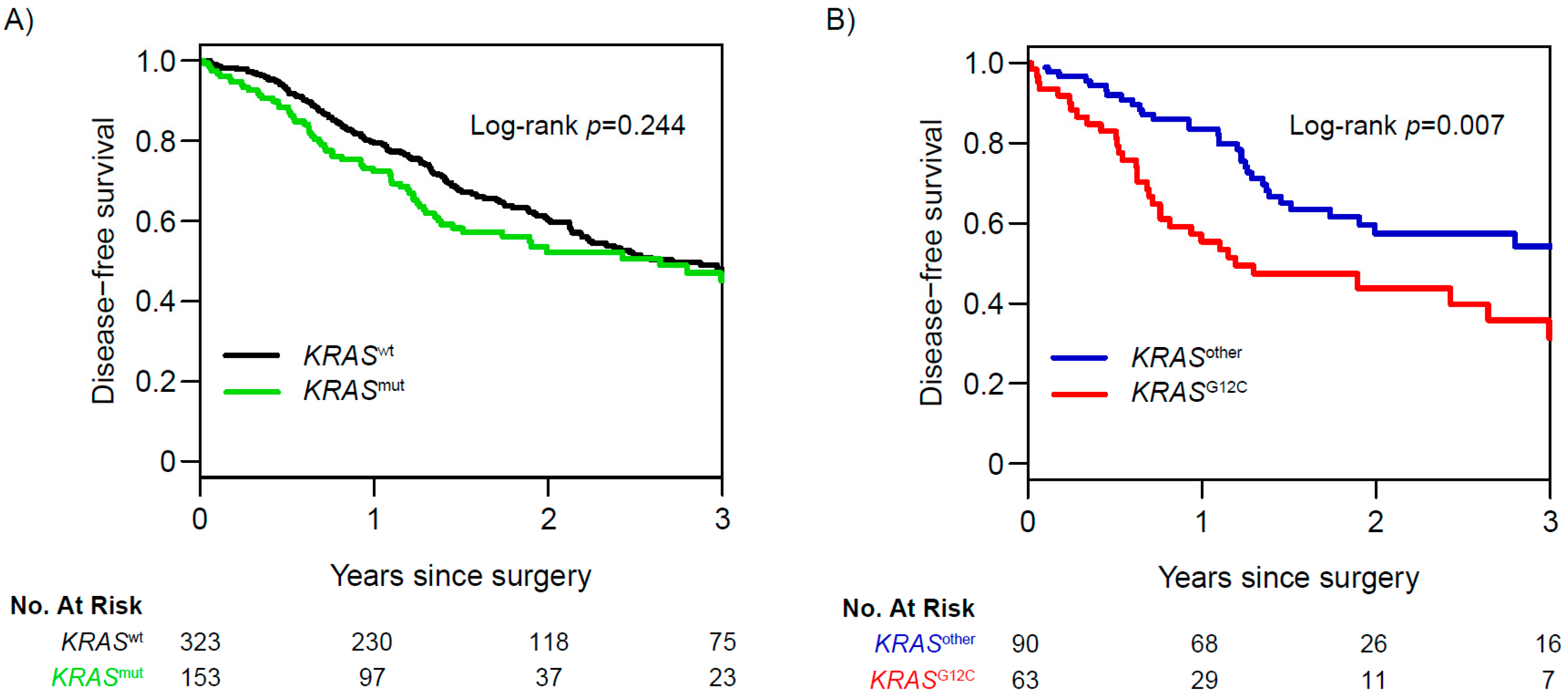
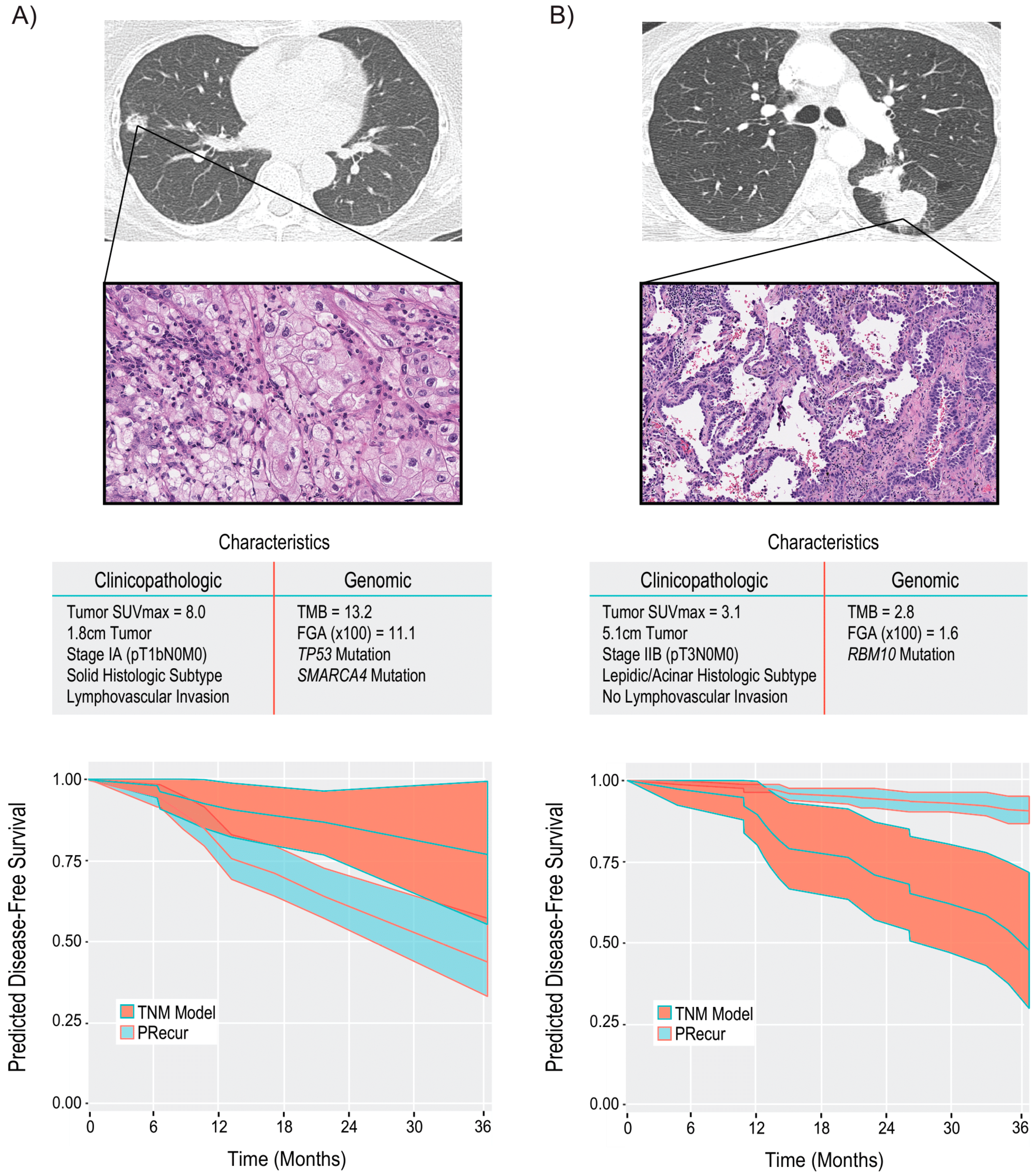
4. NGS for Prognosis and Prediction of Recurrence after Surgery
5. Plasma ctDNA for Early Detection of Lung Cancer and Monitoring for MRD
6. Differentiation between Separate Primary Lung Cancers (SPLCs) and IPMs
7. Use of NGS to Guide Induction and Adjuvant Therapies for Operable Lung Cancer
7.1. Adjuvant Clinical Trials
7.2. Neoadjuvant Clinical Trials
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Mascaux, C.; Tsao, M.S.; Hirsch, F.R. Genomic testing in lung cancer: Past, present, and future. J. Natl. Compr. Canc. Netw. 2018, 16, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.; Black, J.R.M.; Reading, J.L.; Litchfield, K.; Turajlic, S.; McGranahan, N.; Jamal-Hanjani, M.; Swanton, C. Tracking cancer evolution through the disease course. Cancer Discov. 2021, 11, 916–932. [Google Scholar] [CrossRef]
- Jones, G.D.; Brandt, W.S.; Shen, R.; Sanchez-Vega, F.; Tan, K.S.; Martin, A.; Zhou, J.; Berger, M.; Solit, D.B.; Schultz, N.; et al. A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma. JAMA Surg. 2021, 156, e205601. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.J.; Yeh, Y.C.; Jeng, W.J.; Wu, K.J.; Huang, B.S.; Wu, Y.C.; Chou, T.Y.; Hsu, W.H. Predictive value of the international association for the study of lung cancer/American Thoracic Society/European Respiratory Society classification of lung adenocarcinoma in tumor recurrence and patient survival. J. Clin. Oncol. 2014, 32, 2357–2364. [Google Scholar] [CrossRef]
- Warth, A.; Muley, T.; Meister, M.; Stenzinger, A.; Thomas, M.; Schirmacher, P.; Schnabel, P.A.; Budczies, J.; Hoffmann, H.; Weichert, W. The novel histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J. Clin. Oncol. 2012, 30, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H.; Kadota, K.; Chaft, J.E.; Buitrago, D.; Sima, C.S.; Lee, M.C.; Huang, J.; Travis, W.D.; Rizk, N.P.; Rudin, C.M.; et al. Solid predominant histologic subtype in resected stage i lung adenocarcinoma is an independent predictor of early, extrathoracic, multisite recurrence and of poor postrecurrence survival. J. Clin. Oncol. 2015, 33, 2874–2877. [Google Scholar] [CrossRef]
- Motono, N.; Matsui, T.; Machida, Y.; Usuda, K.; Uramoto, H. Prognostic significance of histologic subtype in pStage I lung adenocarcinoma. Med. Oncol. 2017, 34, 100. [Google Scholar] [CrossRef]
- Caso, R.; Sanchez-Vega, F.; Tan, K.S.; Mastrogiacomo, B.; Zhou, J.; Jones, G.D.; Nguyen, B.; Schultz, N.; Connolly, J.G.; Brandt, W.S.; et al. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J. Thorac. Oncol. 2020, 15, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, L.; Guo, L.; Wu, C.; Zhou, J.; Zhou, Y.; Ma, J.; Li, X.; Ji, P.; Wang, M.; et al. Comparative study on the mutational profile of adenocarcinoma and squamous cell carcinoma predominant histologic subtypes in Chinese non-small cell lung cancer patients. Thorac. Cancer 2020, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Kobayashi, M.; Fujimoto, M.; Kawakami, F.; Tsuruyama, T.; Travis, W.D.; Date, H.; Haga, H. Validation of the IASLC/ATS/ERS lung adenocarcinoma classification for prognosis and association with EGFR and KRAS gene mutations: Analysis of 440 Japanese patients. J. Thorac. Oncol. 2013, 8, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Sanchez-Vega, F.; Caso, R.; Tan, K.S.; Brandt, W.S.; Jones, G.D.; Yan, S.; Adusumilli, P.S.; Bott, M.; Huang, J.; et al. Analysis of tumor genomic pathway alterations using broad-panel next-generation sequencing in surgically resected lung adenocarcinoma. Clin. Cancer Res. 2019, 25, 7475–7484. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC enzymes: Mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chong, W.; Teng, C.; Yao, Y.; Wang, X.; Li, X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 2019, 110, 2348–2356. [Google Scholar] [CrossRef]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Brandt, W.S.; Yan, W.; Zhou, J.; Tan, K.S.; Montecalvo, J.; Park, B.J.; Adusumilli, P.S.; Huang, J.; Bott, M.J.; Rusch, V.W.; et al. Outcomes after neoadjuvant or adjuvant chemotherapy for cT2-4N0-1 non-small cell lung cancer: A propensity-matched analysis. J. Thorac. Cardiovasc. Surg. 2019, 157, 743–753. [Google Scholar] [CrossRef]
- Pignon, J.-P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.-Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. In Database of Abstracts of Reviews of Effects (DARE): Quality-Assessed Reviews [Internet]; Centre for Reviews and Dissemination: York, UK, 2008. [Google Scholar]
- Group, N.M.A.C. Preoperative chemotherapy for non-small-cell lung cancer: A systematic review and meta-analysis of individual participant data. Lancet 2014, 383, 1561–1571. [Google Scholar]
- Kim, I.A.; Hur, J.Y.; Kim, H.J.; Park, J.H.; Hwang, J.J.; Lee, S.A.; Lee, S.E.; Kim, W.S.; Lee, K.Y. Targeted next-generation sequencing analysis for recurrence in early-stage lung adenocarcinoma. Ann. Surg. Oncol. 2021, 28, 3983–3993. [Google Scholar] [CrossRef]
- Cho, W.C.S.; Tan, K.T.; Ma, V.W.S.; Li, J.Y.C.; Ngan, R.K.C.; Cheuk, W.; Yip, T.T.C.; Yang, Y.T.; Chen, S.J. Targeted next-generation sequencing reveals recurrence-associated genomic alterations in early-stage non-small cell lung cancer. Oncotarget 2018, 9, 36344–36357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Fang, W.; Li, C.; Tang, K.; Zhang, J.; Lei, Y.; He, W.; Peng, S.; Kuang, M.; Zhang, H. Development and validation of a novel signature to predict overall survival in “driver gene–negative” lung adenocarcinoma (LUAD): Results of a multicenter study. Clin. Cancer Res. 2019, 25, 1546–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.D.; Caso, R.; Tan, K.S.; Mastrogiacomo, B.; Sanchez-Vega, F.; Liu, Y.; Connolly, J.G.; Murciano-Goroff, Y.R.; Bott, M.J.; Adusumilli, P.S. KRASG12C mutation is associated with increased risk of recurrence in surgically resected lung adenocarcinoma. Clin. Cancer Res. 2021, 27, 2604–2612. [Google Scholar] [CrossRef]
- Devarakonda, S.; Rotolo, F.; Tsao, M.S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor mutation burden as a biomarker in resected non-small-cell lung cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef] [PubMed]
- Owada-Ozaki, Y.; Muto, S.; Takagi, H.; Inoue, T.; Watanabe, Y.; Fukuhara, M.; Yamaura, T.; Okabe, N.; Matsumura, Y.; Hasegawa, T.; et al. Prognostic impact of tumor mutation burden in patients with completely resected non-small cell lung cancer: Brief report. J. Thorac. Oncol. 2018, 13, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 2018, 7, e37294. [Google Scholar] [CrossRef]
- Biswas, D.; Birkbak, N.J.; Rosenthal, R.; Hiley, C.T.; Lim, E.L.; Papp, K.; Boeing, S.; Krzystanek, M.; Djureinovic, D.; La Fleur, L.; et al. A clonal expression biomarker associates with lung cancer mortality. Nat. Med. 2019, 25, 1540–1548. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early stage NSCLC—challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef] [Green Version]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Liquid biopsy: From discovery to clinical application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): A statement paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, J.K.; Offin, M.; Stephens, D.; Ni, A.; Lee, A.; Pavlakis, N.; Clarke, S.; Diakos, C.I.; Datta, S.; Tandon, N.; et al. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J. Natl. Cancer Inst. 2019, 111, 575–583. [Google Scholar] [CrossRef]
- Li, B.T.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.S.; Shen, R.; Isbell, J.M.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 2020, 183, 363–376. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nabet, B.Y.; Rizvi, H.; Chaudhuri, A.A.; Wells, D.K.; Dunphy, M.P.S.; Chabon, J.J.; Liu, C.L.; Hui, A.B.; Arbour, K.C.; et al. Circulating tumor DNA analysis to assess risk of progression after long-term response to PD-(L)1 blockade in NSCLC. Clin. Cancer Res. 2020, 26, 2849–2858. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.O.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, N.; Melamed, M.R. Multiple primary lung cancers. J. Thorac. Cardiovasc. Surg. 1975, 70, 606–612. [Google Scholar] [CrossRef]
- Chang, J.C.; Alex, D.; Bott, M.; Tan, K.S.; Seshan, V.; Golden, A.; Sauter, J.L.; Buonocore, D.J.; Vanderbilt, C.M.; Gupta, S. Comprehensive NGS unambiguously distinguishes separate primary lung carcinomas from intra-pulmonary metastases: Comparison with standard histopathologic approach. Clin. Cancer Res. 2019, 25, 7113–7125. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.B.; Kadi, W.; Walts, A.E.; Marchevsky, A.M.; Pao, A.; Aguiluz, A.; Mudalige, T.; Liu, Z.; Deng, N.; Lopategui, J. Next-generation sequencing: A novel approach to distinguish multifocal primary lung adenocarcinomas from intrapulmonary metastases. J. Mol. Diagn. 2017, 19, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Shen, Q.; Mardekian, S.; Solomides, C.; Wang, Z.X.; Evans, N.R., 3rd. Molecular profiling of key driver genes improves staging accuracy in multifocal non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2020, 160, e71–e79. [Google Scholar] [CrossRef]
- Mansuet-Lupo, A.; Barritault, M.; Alifano, M.; Janet-Vendroux, A.; Zarmaev, M.; Biton, J.; Velut, Y.; Le Hay, C.; Cremer, I.; Regnard, J.F.; et al. Proposal for a combined histomolecular algorithm to distinguish multiple primary adenocarcinomas from intrapulmonary metastasis in patients with multiple lung tumors. J. Thorac. Oncol. 2019, 14, 844–856. [Google Scholar] [CrossRef]
- Murphy, S.J.; Harris, F.R.; Kosari, F.; Terra, S.B.S.P.; Nasir, A.; Johnson, S.H.; Serla, V.; Smadbeck, J.B.; Halling, G.C.; Karagouga, G.; et al. Using genomics to differentiate multiple primaries from metastatic lung cancer. J. Thorac. Oncol. 2019, 14, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Vincenten, J.P.; van Essen, H.F.; Lissenberg-Witte, B.I.; Bulkmans, N.W.; Krijgsman, O.; Sie, D.; Eijk, P.P.; Smit, E.F.; Ylstra, B.; Thunnissen, E. Clonality analysis of pulmonary tumors by genome-wide copy number profiling. PLoS ONE 2019, 14, e0223827. [Google Scholar]
- Corsini, E.M.; Wang, J.; Wu, C.C.; Fujimoto, J.; Negrao, M.V.; Chen, R.; Quek, K.; Mitchell, K.G.; Chow, C.B.; Little, L.; et al. Genomic assessment distinguishes intrapulmonary metastases from synchronous primary lung cancers. J. Thorac. Dis. 2020, 12, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Addeo, A.; Russo, A.; Gregorc, V.; Cortinovis, D.; Rolfo, C.D. Targeted therapies in early stage NSCLC: Hype or hope? Int. J. Mol. Sci. 2020, 21, 6329. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Graham, R.P.; Treece, A.L.; Lindeman, N.I.; Vasalos, P.; Shan, M.; Jennings, L.J.; Rimm, D.L. Worldwide frequency of commonly detected EGFR mutations. Arch. Pathol. Lab. Med. 2018, 142, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Chudgar, N.P.; Jones, D.R. Thoracoscopic lobectomy following neoadjuvant tyrosine kinase inhibitor treatment. JTCVS Tech. 2021, 7, 294–297. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detection Assay | Developer | Application |
|---|---|---|
| CAPP-Seq | Stanford | MRD |
| Avenio | Roche | MRD |
| TEC-Seq | Johns Hopkins | MRD |
| Lung-CLiP | Stanford | Early detection |
| Cancer SEEK | Thrive/Exact Sciences | Early detection |
| Galleri | GRAIL | Early detection |
| Delfi | Delfi Diagnostics | Early detection |
| Signatera | Natera | MRD |
| PCM | ArcherDX | MRD |
| RaDaR | Inivata | MRD |
| PhasED-Seq | Foresight Diagnostics | MRD |
| Name/NCT# | Trial Type | Genomic Target | Phase | Primary Outcome | No. of Patients | Population/Trial Design | Completion Date |
|---|---|---|---|---|---|---|---|
| ALCHEMIST/NCT02201992 | Adjuvant | ALK | III-R | OS | 168 | Pathologic stage IB–IIIA/crizotinib × 24 months vs. placebo | 2022 |
| ALCHEMIST/NCT02193282 | Adjuvant | EGFR | III-R | OS | 450 | Pathologic stage IB–IIIA/erlotinib × 24 months vs. placebo | 2021 |
| ALINA/NCT03456076 | Adjuvant | ALK | III-R | DFS | 255 | Pathologic stage IB–IIIA/alectinib × 24 months vs. chemotherapy | 2026 |
| MERMAID 1/NCT04385368 | Adjuvant | ctDNA | III-R | DFS | 332 | Pathologic stage II or III/durvalumab + SOC chemotherapy vs SOC chemotherapy | 2026 |
| MERMAID 2/NCT04642469 | Adjuvant | ctDNA | III-R | DFS | 284 | Pathologic stage II or III/durvalumab vs placebo | 2027 |
| NEOADAURA/NCT04351555 | Neoadjuvant | EGFR | III-R | MPR | 328 | Clinical stage II or III/SOC chemotherapy vs osimertinib + SOC chemotherapy vs osimertinib alone | 2024 |
| LEADER/NCT04712877 | Neoadjuvant | Multiple | II | Feasibility | 1000 | Clinical stage IB–IIIA/tumor DNA for NGS before surgery | 2026 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lengel, H.B.; Connolly, J.G.; Jones, G.D.; Caso, R.; Zhou, J.; Sanchez-Vega, F.; Mastrogiacomo, B.; Isbell, J.M.; Li, B.T.; Liu, Y.; et al. The Emerging Importance of Tumor Genomics in Operable Non-Small Cell Lung Cancer. Cancers 2021, 13, 3656. https://doi.org/10.3390/cancers13153656
Lengel HB, Connolly JG, Jones GD, Caso R, Zhou J, Sanchez-Vega F, Mastrogiacomo B, Isbell JM, Li BT, Liu Y, et al. The Emerging Importance of Tumor Genomics in Operable Non-Small Cell Lung Cancer. Cancers. 2021; 13(15):3656. https://doi.org/10.3390/cancers13153656
Chicago/Turabian StyleLengel, Harry B., James G. Connolly, Gregory D. Jones, Raul Caso, Jian Zhou, Francisco Sanchez-Vega, Brooke Mastrogiacomo, James M. Isbell, Bob T. Li, Yuan Liu, and et al. 2021. "The Emerging Importance of Tumor Genomics in Operable Non-Small Cell Lung Cancer" Cancers 13, no. 15: 3656. https://doi.org/10.3390/cancers13153656
APA StyleLengel, H. B., Connolly, J. G., Jones, G. D., Caso, R., Zhou, J., Sanchez-Vega, F., Mastrogiacomo, B., Isbell, J. M., Li, B. T., Liu, Y., Rekhtman, N., & Jones, D. R. (2021). The Emerging Importance of Tumor Genomics in Operable Non-Small Cell Lung Cancer. Cancers, 13(15), 3656. https://doi.org/10.3390/cancers13153656