
Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Cellular Components and Molecules Associated with an Inhibitory Tumor Immune Microenvironment
2.1. Regulatory T-Cells
2.2. Myeloid-Derived Suppresor Cells
2.3. Tumor-Associated Macrophages
2.4. Cancer-Associated Fibroblasts and Vascular Endothelial Cells
2.5. Other Stromal Cells
2.6. Immunosuppressive Metabolites
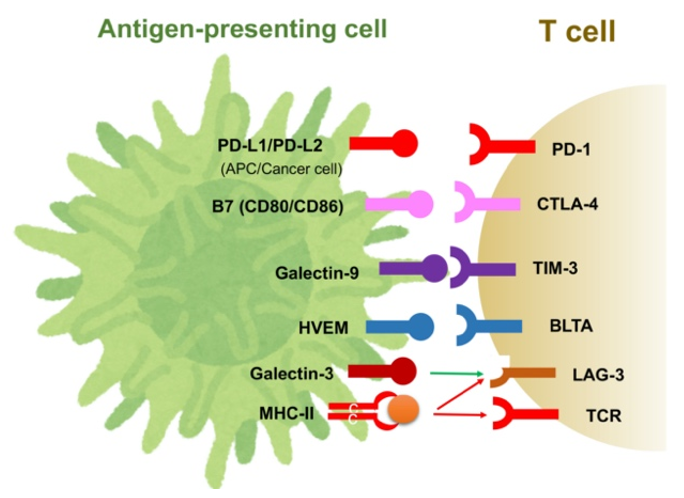
2.7. Immune Checkpoint Molecules
3. Unique Aspects of Immunological Characteristics in the Liver and Hepatocarcinogenesis
4. Signaling Pathways and the Immune Microenvironment of Tumors
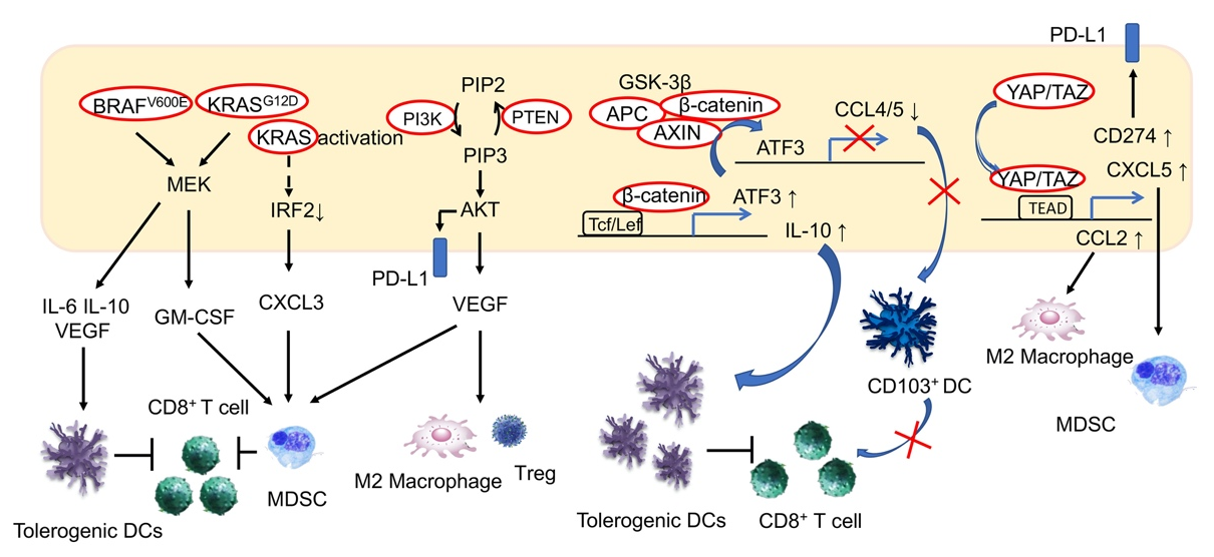
4.1. RAS/MAPK Signaling Pathway
4.2. PI3K/Akt Signaling Pathway
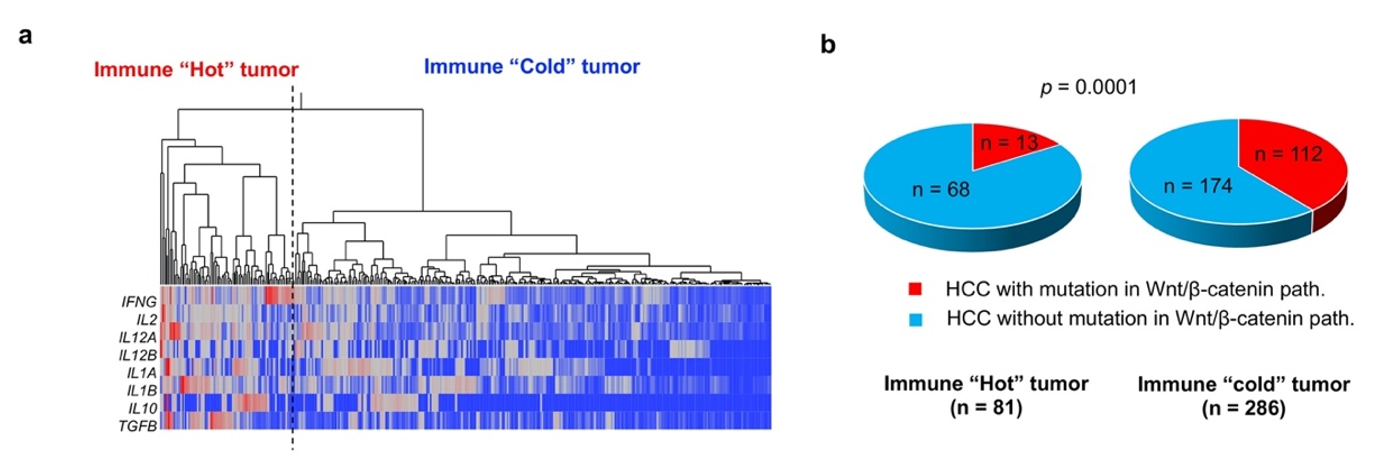
4.3. Wnt/β-Catenin Signaling Pathway
4.4. MYC Gene
4.5. Chromatin Remodeling Pathway
4.6. JAK/STAT Signaling Pathway
4.7. Hippo Signaling Pathway
4.8. DNA Repair Pathway
4.9. VEGF Signaling
5. Signaling Pathway Abnormalities and the Immune Microenvironment in HCC
6. Conclusions
Funding
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Recent Advances in Systemic Therapy for Hepatocellular Carcinoma in an Aging Society: 2020 Update. Liver Cancer 2020, 9, 640–662. [Google Scholar] [CrossRef]
- Nishida, N.; Arizumi, T.; Hagiwara, S.; Ida, H.; Sakurai, T.; Kudo, M. MicroRNAs for the Prediction of Early Response to Sorafenib Treatment in Human Hepatocellular Carcinoma. Liver Cancer 2017, 6, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers 2018, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Kudo, M. Immune checkpoint blockade for the treatment of human hepatocellular carcinoma. Hepatol. Res. 2018, 48, 622–634. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers 2020, 12, 1274. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Role of Immune Checkpoint Blockade in the Treatment for Human Hepatocellular Carcinoma. Dig. Dis. 2017, 35, 618–622. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Immunological Microenvironment of Hepatocellular Carcinoma and Its Clinical Implication. Oncology 2017, 92 (Suppl. 1), 40–49. [Google Scholar] [CrossRef]
- Aoki, T.; Kudo, M.; Ueshima, K.; Morita, M.; Chishina, H.; Takita, M.; Hagiwara, S.; Ida, H.; Minami, Y.; Tsurusaki, M.; et al. Exploratory Analysis of Lenvatinib Therapy in Patients with Unresectable Hepatocellular Carcinoma Who Have Failed Prior PD-1/PD-L1 Checkpoint Blockade. Cancers 2020, 12, 3048. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N. Clinical implications of the dual blockade of the PD-1/PD-L1 and vascular endothelial growth factor axes in the treatment of hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2020, 9, 640–643. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Oncogenic Signal and Tumor Microenvironment in Hepatocellular Carcinoma. Oncology 2017, 93 (Suppl. 1), 160–164. [Google Scholar] [CrossRef]
- Sprinzl, M.F.; Galle, P.R. Immune control in hepatocellular carcinoma development and progression: Role of stromal cells. Semin. Liver Dis. 2014, 34, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yamaguchi, R.; Hasegawa, T.; Shimada, S.; Arihiro, K.; Hayashi, S.; Maejima, K.; Nakano, K.; Fujimoto, A.; Ono, A.; et al. Classification of primary liver cancer with immunosuppression mechanisms and correlation with genomic alterations. EBioMedicine 2020, 53, 102659. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.; Grone, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Arihara, F.; Mizukoshi, E.; Kitahara, M.; Takata, Y.; Arai, K.; Yamashita, T.; Nakamoto, Y.; Kaneko, S. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol. Immunother 2013, 62, 1421–1430. [Google Scholar] [CrossRef] [Green Version]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Neaud, V.; Faouzi, S.; Guirouilh, J.; Le Bail, B.; Balabaud, C.; Bioulac-Sage, P.; Rosenbaum, J. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: Evidence for a role of hepatocyte growth factor. Hepatology 1997, 26, 1458–1466. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochst, B.; Schildberg, F.A.; Sauerborn, P.; Gabel, Y.A.; Gevensleben, H.; Goltz, D.; Heukamp, L.C.; Turler, A.; Ballmaier, M.; Gieseke, F.; et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 2013, 59, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Kuang, D.M.; Wu, Y.; Xiao, X.; Li, X.F.; Li, T.J.; Zheng, L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J. Immunol. 2012, 188, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef]
- Nishida, N.; Sakai, K.; Morita, M.; Aoki, T.; Takita, M.; Hagiwara, S.; Komeda, Y.; Takenaka, M.; Minami, Y.; Ida, H.; et al. Association between Genetic and Immunological Background of Hepatocellular Carcinoma and Expression of Programmed Cell Death-1. Liver Cancer 2020, 9, 426–439. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Sarobe, P.; Hervas-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Schinzari, V.; Timperi, E.; Pecora, G.; Palmucci, F.; Gallerano, D.; Grimaldi, A.; Covino, D.A.; Guglielmo, N.; Melandro, F.; Manzi, E.; et al. Wnt3a/beta-Catenin Signaling Conditions Differentiation of Partially Exhausted T-effector Cells in Human Cancers. Cancer Immunol. Res. 2018, 6, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Pacella, I.; Cammarata, I.; Focaccetti, C.; Miacci, S.; Gulino, A.; Tripodo, C.; Rava, M.; Barnaba, V.; Piconese, S. Wnt3a Neutralization Enhances T-cell Responses through Indirect Mechanisms and Restrains Tumor Growth. Cancer Immunol. Res. 2018, 6, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, D.; Paroli, M.; Francavilla, V.; Videtta, M.; Morrone, S.; Labbadia, G.; Cerino, A.; Mondelli, M.U.; Barnaba, V. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J. Clin. Investig. 2009, 119, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.G.; Kim, C.; Yoon, S.E.; Kim, K.H.; Choi, S.J.; Kang, B.; Kim, H.R.; Park, S.H.; Shin, E.C.; Kim, Y.Y.; et al. Hyperprogressive disease during PD-1 blockade in patients with advanced hepatocellular carcinoma. J. Hepatol. 2021, 74, 350–359. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J. Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Romero, J.M.; Jimenez, P.; Cabrera, T.; Cozar, J.M.; Pedrinaci, S.; Tallada, M.; Garrido, F.; Ruiz-Cabello, F. Coordinated downregulation of the antigen presentation machinery and HLA class I/beta2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer 2005, 113, 605–610. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, M. Evaluation of BRCA1 and BRCA2 as Indicators of Response to Immune Checkpoint Inhibitors. JAMA Netw. Open 2021, 4, e217728. [Google Scholar] [CrossRef]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Nishida, N.; Sakai, K.; Aoki, T.; Chishina, H.; Takita, M.; Ida, H.; Hagiwara, S.; Minami, Y.; Ueshima, K.; et al. Immunological microenvironment predicts the survival of the patients with hepatocellular carcinoma treated with anti-PD-1 antibody. Liver Cancer 2021, 10, 380–393, in press. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NCT Number 1 (Trial Name) | MTAs/ICIs 2 | Targets of MTAs | Setting |
|---|---|---|---|
| Phase I/II | |||
| NCT03299946 (CaboNivo) | Cabozantinib/ Nivolumab | TKI for VEGFR2, MET, AXL, etc. | neoadjuvant |
| NCT03170960 (COSMIC-021) | Cabozantinib/ Atezolizumab | Same as above | First-line |
| NCT04442581 | Cabozantinib/ Pembrolizumab | Same as above | First-line |
| NCT01658878 (CheckMate 040) | Cabozantinib/Nivolumab±Ipilimumab | Same as above | First -and second-line |
| NCT03289533 (VEGF Liver 100) | Axitinib/ Avelumab | TKI for VEGFR1-3, PDGFR, c-kit, etc. | First-line and AFP ≥ 400 ng/mL |
| NCT03841201, NCT03418922 | Lenvatinib/ Nivolumab | TKI for VEGFR1-3, FGFR1-4, etc. | First-line |
| NCT03347292 (Bayer 19497) | Regorafenib/Pembrolizumab | TKI for VEGFR1-3, TIE2, PDGFR, c-kit, RET, etc. | First-line |
| NCT04310709 (RENOBATE) | Regorafenib/Nivolumab | Same as above | First-line |
| NCT04183088 | Regorafenib/Tislelizumab | Same as above | First-line |
| NCT03941873 | Sitravatinib/Tislelizumab | TKI for VEGFR2, c-kit, AXL, etc. | First-line and later |
| NCT03475953 (REGOMUNE) | Regorafenib/Avelumab | TKI for VEGFR1-3, TIE2, PDGFR, c-kit, RET, etc. | Second-line |
| NCT04170556 (GOING) | Regorafenib/Followed by Nivolumab | Same as above | Second-line |
| NCT03539822 (CAMILLA) | Cabozantinib/Durvalumab | TKI for VEGFR2, MET, AXL, etc. | Second-line |
| NCT02572687 | Ramucirumab/Durvalumab | Ab for VEGFR2 | Second-line and AFP ≥ 1.5x ULN |
| NCT02423343 | Galunisertib/Nivolumab | TKI for TGβR1 | Second-line and AFP ≥ 200 ng/mL |
| Phase III | |||
| NCT04102098 (IMbrave050) | Bevacizumab/Atezolizumab | Ab for VEGFA | Adjuvant |
| NCT03847428 (EMERALD-2) | Bevacizumab/±Durvalumab (vs. placebo) | Same as above | Adjuvant |
| NCT03713593 (LEAP-002) | Lenvatinib/Pembrolizumab (vs. Lenvatinib) | TKI for VEGFR1-3, FGFR1-4, etc. | First-line |
| NCT03755791 (COSMIC-312) | Cabozantinib/Atezolizumab (vs.orafenib or. Cabozantinib) | TKI for VEGFR2,MET, AXL, etc. | First-line |
| NCT03764293 | Apatinib/Camrelizumab (vs. sorafenib) | TKI for VEGFR2 | First-line |
| Oncogenic Pathway | Mutation | CD8+ TILs | p Value 2 | |
|---|---|---|---|---|
| Median1 | 25–75th Percentile | |||
| Wnt/β-catenin path. | with | 6.18 | 1.30–26.9 | 0.0082 |
| without | 17.6 | 5.77–38.0 | ||
| p53/cell cycle path. | with | 18.7 | 5.70–32.7 | 0.7505 |
| without | 12.9 | 3.60–38.3 | ||
| PI3K/Akt path. | with | 1.14 | 0.17–2.03 | 0.5836 |
| without | 1.16 | 0.36–2.88 | ||
| Chromatin remodeling | with | 17.3 | 2.16–31.1 | 0.8056 |
| without | 14.1 | 4.44–36.0 | ||
| Epigenetic regulator | with | 0.75 | 0.15–1.81 | 0.1488 |
| without | 1.29 | 0.42–2.88 | ||
| Oxidative/ER stress | with | 1.63 | 0.53–5.74 | 0.1871 |
| without | 1.11 | 0.28–2.72 | ||
| DNA repair | with | 1.24 | 0.45–2.97 | 0.7392 |
| without | 1.14 | 0.28–2.73 | ||
| TERT promoter | with | 1.40 | 0.38–2.81 | 0.5093 |
| without | 1.03 | 0.28–2.70 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, N. Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers 2021, 13, 3666. https://doi.org/10.3390/cancers13153666
Nishida N. Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers. 2021; 13(15):3666. https://doi.org/10.3390/cancers13153666
Chicago/Turabian StyleNishida, Naoshi. 2021. "Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma" Cancers 13, no. 15: 3666. https://doi.org/10.3390/cancers13153666
APA StyleNishida, N. (2021). Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers, 13(15), 3666. https://doi.org/10.3390/cancers13153666