
DC-Derived Exosomes for Cancer Immunotherapy


Abstract
:Simple Summary
Abstract
1. Dendritic Cells, Anti-Tumor Immunity and Dendritic Cell-Based Cancer Vaccines
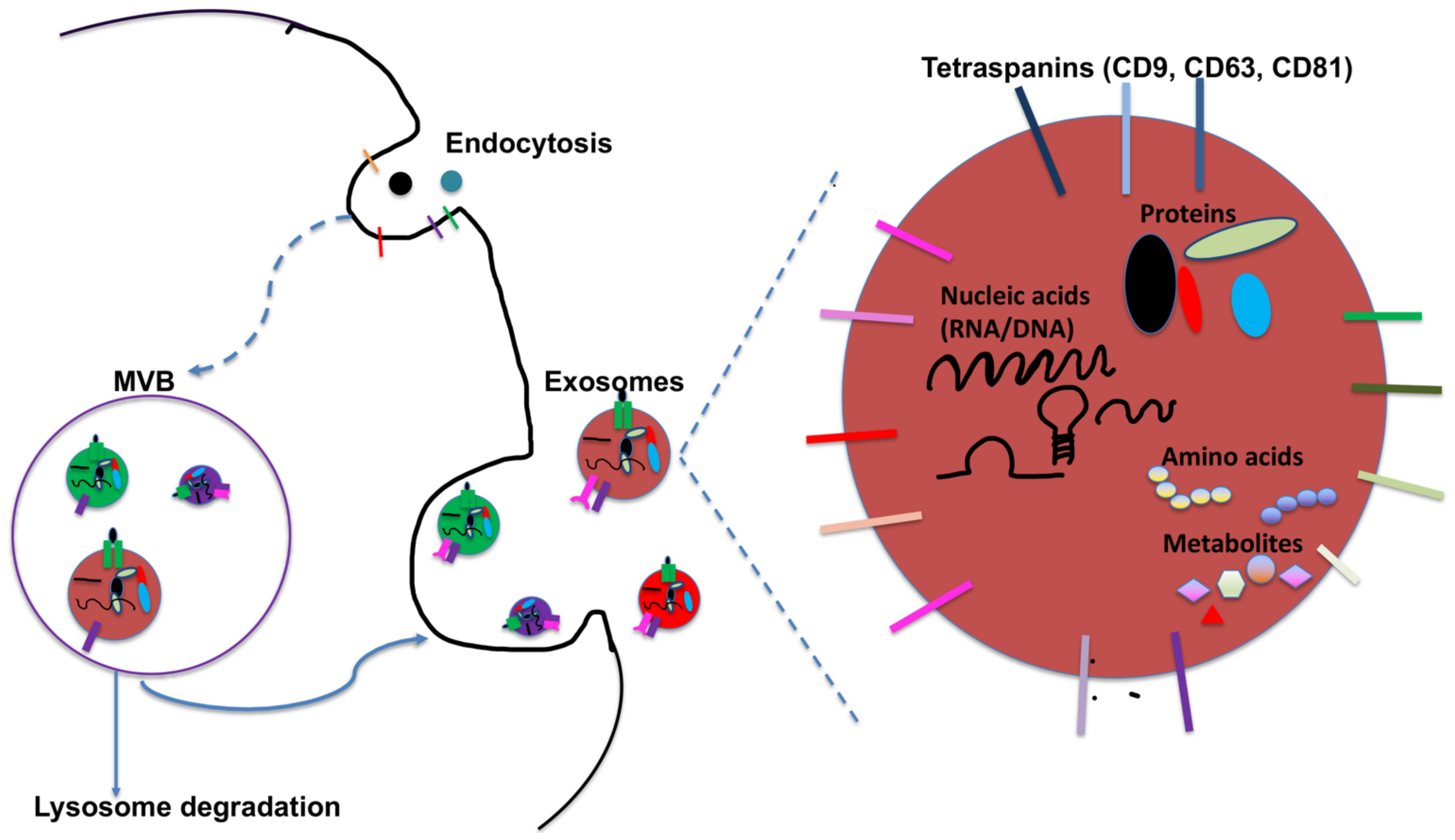
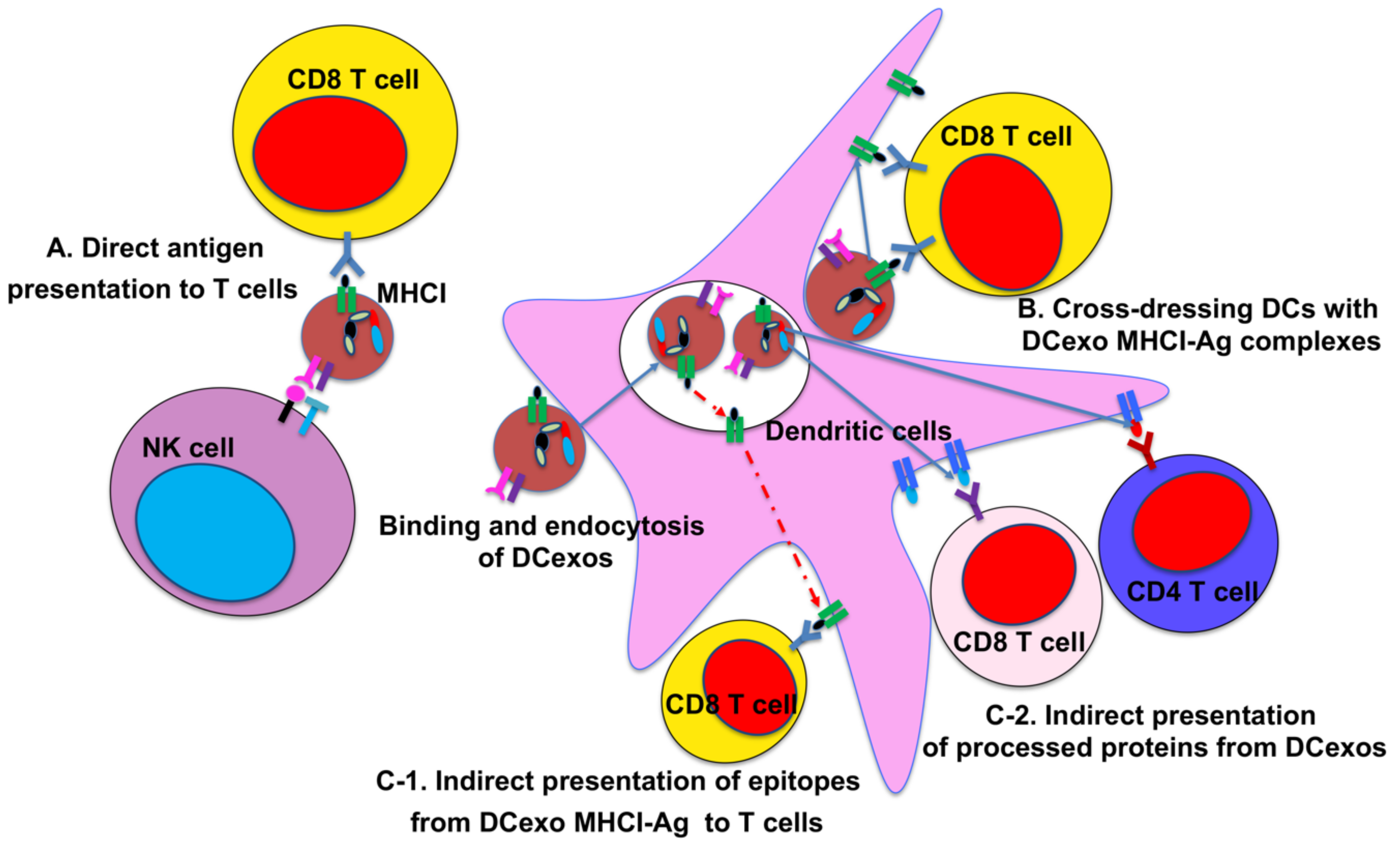
2. DC-Derived Exosomes and Their Function
3. DC-Derived Exosomes in Clinical Trials
4. DCexo Phase I Clinical Trials
5. DCexo Phase II Clinical Trial
6. Conclusions and Future Directions
7. Plasmacytoid DC-Derived Exosomes—The New Addition to DCexos
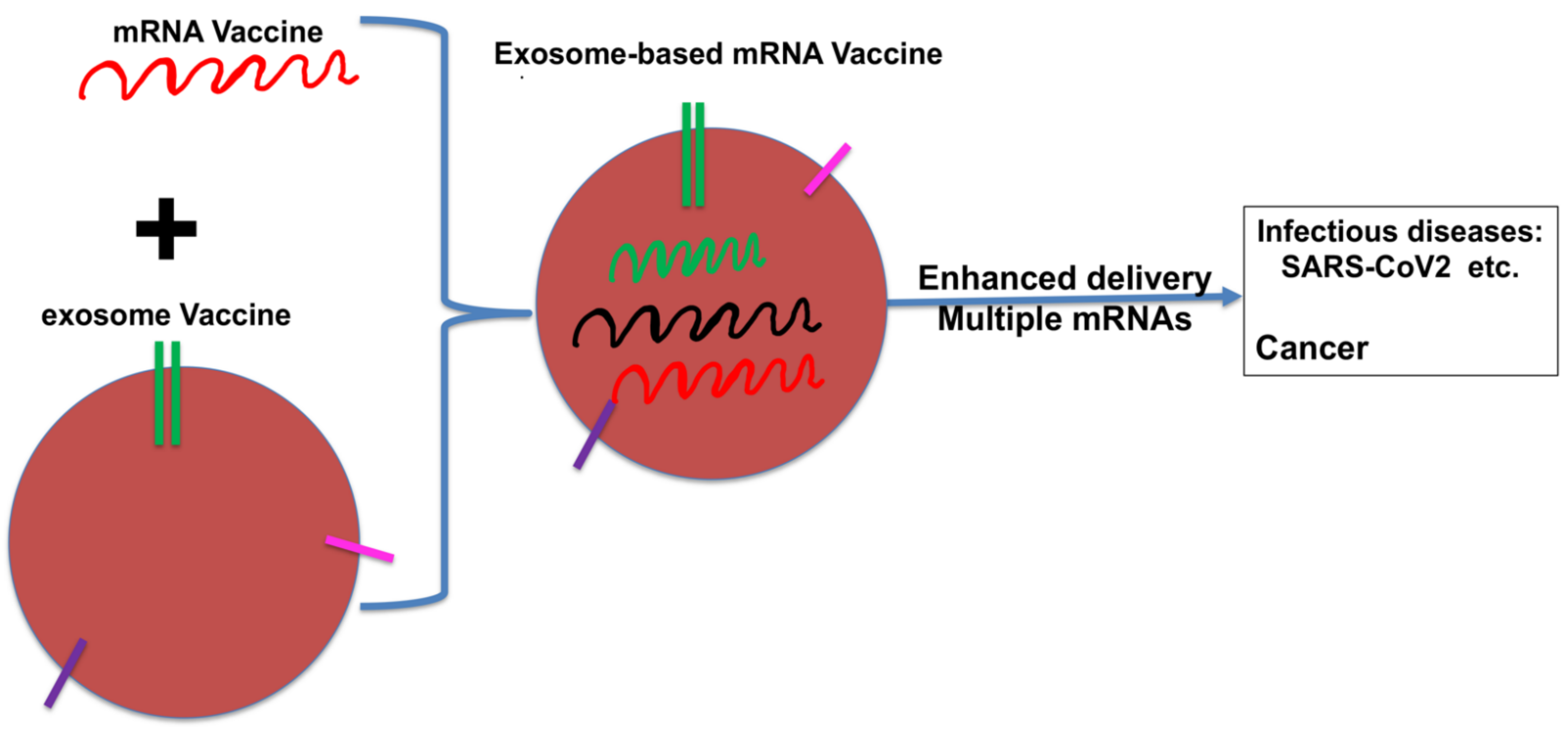
8. The Future of DCexos as Cancer Vaccines?
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, A.; Dammeijer, F.; Aerts, J.; Vroman, H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for in vitro Antigen Loading of Different DC Subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.A., 3rd; Dutertre, C.A.; Ginhoux, F.; Murphy, K.M. Genetic models of human and mouse dendritic cell development and function. Nat. Rev. Immunol. 2021, 21, 101–115. [Google Scholar] [CrossRef]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2018. [Google Scholar] [CrossRef]
- Chrisikos, T.T.; Zhou, Y.; Slone, N.; Babcock, R.; Watowich, S.S.; Li, H.S. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol. Immunol. 2019, 110, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Gutierrez-Martinez, E.; Planes, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Honma, K.; Matsuyama, T.; Suzuki, K.; Toriyama, K.; Akitoyo, I.; Yamamoto, K.; Suematsu, T.; Nakamura, M.; Yui, K.; et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc. Natl. Acad. Sci. USA 2004, 101, 8981–8986. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Reizis, B.; Colonna, M.; Trinchieri, G.; Barrat, F.; Gilliet, M. Plasmacytoid dendritic cells: One-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 2011, 11, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid dendritic cell in immunity and cancer. J. Neuroimmunol. 2018, 322, 63–73. [Google Scholar] [CrossRef]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; den Hollander, N.S.; Kant, S.G.; et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, P.F.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.J. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 1976, 117, 2233–2238. [Google Scholar]
- Kurts, C.; Robinson, B.W.; Knolle, P.A. Cross-priming in health and disease. Nat. Rev. Immunol. 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Ohlfest, J.R. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett. 2012, 325, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fabian, K.L.; Taylor, J.L.; Storkus, W.J. Therapeutic Use of Dendritic Cells to Promote the Extranodal Priming of Anti-Tumor Immunity. Front. Immunol. 2013, 4, 388. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Mitchell, D.A. The development of dendritic cell vaccine-based immunotherapies for glioblastoma. Semin Immunopathol. 2017, 39, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.S.; Jiang, A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines 2020, 8, 706. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii44–xii55. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Pitt, J.M.; Andre, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Invest. 2016, 126, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother Cancer 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Marar, C.; Starich, B.; Wirtz, D. Extracellular vesicles in immunomodulation and tumor progression. Nat. Immunol. 2021, 22, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Stefanius, K.; Servage, K.; Orth, K. Exosomes in cancer development. Curr. Opin. Genet. Dev. 2021, 66, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Leone, D.A.; Rees, A.J.; Kain, R. Dendritic cells and routing cargo into exosomes. Immunol. Cell Biol. 2018, 96, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Jella, K.K.; Nasti, T.H.; Li, Z.; Malla, S.R.; Buchwald, Z.S.; Khan, M.K. Exosomes, Their Biogenesis and Role in Inter-Cellular Communication, Tumor Microenvironment and Cancer Immunotherapy. Vaccines 2018, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.; Kosgodage, U.; Lange, S.; Inal, J.M. The emerging role of exosome and microvesicle- (EMV-) based cancer therapeutics and immunotherapy. Int. J. Cancer 2017, 141, 428–436. [Google Scholar] [CrossRef]
- Veerman, R.E.; Gucluler Akpinar, G.; Eldh, M.; Gabrielsson, S. Immune Cell-Derived Extracellular Vesicles-Functions and Therapeutic Applications. Trends Mol. Med. 2019. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur. J. Immunol. 2006, 36, 1772–1781. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Segura, E.; Amigorena, S.; Thery, C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol. Dis. 2005, 35, 89–93. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Samuel, M.; Gabrielsson, S. Personalized medicine and back-allogeneic exosomes for cancer immunotherapy. J. Intern. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qazi, K.R.; Gehrmann, U.; Domange Jordo, E.; Karlsson, M.C.; Gabrielsson, S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood 2009, 113, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naslund, T.I.; Gehrmann, U.; Qazi, K.R.; Karlsson, M.C.; Gabrielsson, S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J. Immunol. 2013, 190, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Larssen, P.; Eldh, M.; Martinez-Bravo, M.J.; Wagner, A.K.; Karlsson, M.C.; Gabrielsson, S. Exosomal cancer immunotherapy is independent of MHC molecules on exosomes. Oncotarget 2016, 7, 38707–38717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; Andre, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Munich, S.; Sobo-Vujanovic, A.; Buchser, W.J.; Beer-Stolz, D.; Vujanovic, N.L. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology 2012, 1, 1074–1083. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Delgado, I.; Calzada-Fraile, D.; Sanchez-Madrid, F. Immune Regulation by Dendritic Cell Extracellular Vesicles in Cancer Immunotherapy and Vaccines. Cancers 2020, 12, 3558. [Google Scholar] [CrossRef] [PubMed]
- Bell, B.M.; Kirk, I.D.; Hiltbrunner, S.; Gabrielsson, S.; Bultema, J.J. Designer exosomes as next-generation cancer immunotherapy. Nanomedicine 2016, 12, 163–169. [Google Scholar] [CrossRef]
- Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Ferrari, P.; Biava, P.M. Exosomes and Cell Communication: From Tumour-Derived Exosomes and Their Role in Tumour Progression to the Use of Exosomal Cargo for Cancer Treatment. Cancers 2021, 13, 822. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Escudier, B.; Angevin, E.; Tursz, T.; Zitvogel, L. Exosomes for cancer immunotherapy. Ann. Oncol. 2004, 15 (Suppl 4), iv141–iv144. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M. Dendritic cell extracellular vesicles. Int. Rev. Cell Mol. Biol. 2019, 349, 213–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic potential of extracellular vesicles. Front. Immunol. 2014, 5, 518. [Google Scholar] [CrossRef] [Green Version]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. FEBS J. 2021, 288, 10–35. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Pogge von Strandmann, E.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Boll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Simhadri, V.R.; Reiners, K.S.; Hansen, H.P.; Topolar, D.; Simhadri, V.L.; Nohroudi, K.; Kufer, T.A.; Engert, A.; Pogge von Strandmann, E. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS ONE 2008, 3, e3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Dominiak, A.; Zolnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-Derived Exosomes in Immunosuppression and Immunotherapy. J. Immunol. Res. 2020, 2020, 6272498. [Google Scholar] [CrossRef]
- Segura, E.; Nicco, C.; Lombard, B.; Veron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Thery, C. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Ploix, S.; Lapierre, V.; Thery, C.; Commere, P.H.; Tramalloni, D.; Gorrichon, K.; Virault-Rocroy, P.; Tursz, T.; Lantz, O.; et al. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: A critical role of interferon-gamma. J. Immunother. 2011, 34, 65–75. [Google Scholar] [CrossRef]
- Taieb, J.; Chaput, N.; Schartz, N.; Roux, S.; Novault, S.; Menard, C.; Ghiringhelli, F.; Terme, M.; Carpentier, A.F.; Darrasse-Jeze, G.; et al. Chemoimmunotherapy of tumors: Cyclophosphamide synergizes with exosome based vaccines. J. Immunol. 2006, 176, 2722–2729. [Google Scholar] [CrossRef]
- Viaud, S.; Flament, C.; Zoubir, M.; Pautier, P.; LeCesne, A.; Ribrag, V.; Soria, J.C.; Marty, V.; Vielh, P.; Robert, C.; et al. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res. 2011, 71, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Naseri, M.; Bozorgmehr, M.; Zoller, M.; Ranaei Pirmardan, E.; Madjd, Z. Tumor-derived exosomes: The next generation of promising cell-free vaccines in cancer immunotherapy. Oncoimmunology 2020, 9, 1779991. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.; Oshchepkova, A.; Mironova, N. Immunotherapy Based on Dendritic Cell-Targeted/-Derived Extracellular Vesicles-A Novel Strategy for Enhancement of the Anti-tumor Immune Response. Front. Pharmacol. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Nikfarjam, S.; Rezaie, J.; Kashanchi, F.; Jafari, R. Dexosomes as a cell-free vaccine for cancer immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 258. [Google Scholar] [CrossRef]
- Strauss-Albee, D.M.; Fukuyama, J.; Liang, E.C.; Yao, Y.; Jarrell, J.A.; Drake, A.L.; Kinuthia, J.; Montgomery, R.R.; John-Stewart, G.; Holmes, S.; et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci. Transl. Med. 2015, 7, 297ra115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A.; et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013, 19, 4079–4091. [Google Scholar] [CrossRef] [Green Version]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vely, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra255. [Google Scholar] [CrossRef] [PubMed]
- Damo, M.; Wilson, D.S.; Simeoni, E.; Hubbell, J.A. TLR-3 stimulation improves anti-tumor immunity elicited by dendritic cell exosome-based vaccines in a murine model of melanoma. Sci. Rep. 2015, 5, 17622. [Google Scholar] [CrossRef] [Green Version]
- Wimmers, F.; Schreibelt, G.; Skold, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef]
- Davis, I.D.; Chen, Q.; Morris, L.; Quirk, J.; Stanley, M.; Tavarnesi, M.L.; Parente, P.; Cavicchiolo, T.; Hopkins, W.; Jackson, H.; et al. Blood dendritic cells generated with Flt3 ligand and CD40 ligand prime CD8+ T cells efficiently in cancer patients. J. Immunother. 2006, 29, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, I.D.; Quirk, J.; Morris, L.; Seddon, L.; Tai, T.Y.; Whitty, G.; Cavicchiolo, T.; Ebert, L.; Jackson, H.; Browning, J.; et al. A pilot study of peripheral blood BDCA-1 (CD1c) positive dendritic cells pulsed with NY-ESO-1 ISCOMATRIX adjuvant. Immunotherapy 2017, 9, 249–259. [Google Scholar] [CrossRef]
- Hsu, J.L.; Bryant, C.E.; Papadimitrious, M.S.; Kong, B.; Gasiorowski, R.E.; Orellana, D.; McGuire, H.M.; Groth, B.F.S.; Joshua, D.E.; Ho, P.J.; et al. A blood dendritic cell vaccine for acute myeloid leukemia expands anti-tumor T cell responses at remission. Oncoimmunology 2018, 7, e1419114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, J.; Chaperot, L.; Hannani, D.; Bruder Costa, J.; Templier, I.; Trabelsi, S.; Gil, H.; Moisan, A.; Persoons, V.; Hegelhofer, H.; et al. An innovative plasmacytoid dendritic cell line-based cancer vaccine primes and expands antitumor T-cells in melanoma patients in a first-in-human trial. Oncoimmunology 2020, 9, 1738812. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Li, W. Dendritic cell-derived exosomes for cancer immunotherapy: Hope and challenges. Ann. Transl. Med. 2017, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid dendritic cells cross-prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc. Natl Acad Sci. USA 2020. [Google Scholar] [CrossRef]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2013, 93, 343–352. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Charles, J.; Plumas, J. Melanoma hijacks plasmacytoid dendritic cells to promote its own progression. Oncoimmunology 2014, 3, e27402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.J.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspord, C.; Leccia, M.T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A(*)0201(+) plasmacytoid dendritic cells provide a cell-based immunotherapy for melanoma patients. J. Invest. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef]
- Thomas, M.; Ponce-Aix, S.; Navarro, A.; Riera-Knorrenschild, J.; Schmidt, M.; Wiegert, E.; Kapp, K.; Wittig, B.; Mauri, C.; Domine Gomez, M.; et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: Results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann. Oncol. 2018, 29, 2076–2084. [Google Scholar] [CrossRef]
- Nierkens, S.; den Brok, M.H.; Garcia, Z.; Togher, S.; Wagenaars, J.; Wassink, M.; Boon, L.; Ruers, T.J.; Figdor, C.G.; Schoenberger, S.P.; et al. Immune adjuvant efficacy of CpG oligonucleotide in cancer treatment is founded specifically upon TLR9 function in plasmacytoid dendritic cells. Cancer Res. 2011, 71, 6428–6437. [Google Scholar] [CrossRef] [Green Version]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Nierkens, S.; Tel, J.; Janssen, E.; Adema, G.J. Antigen cross-presentation by dendritic cell subsets: One general or all sergeants? Trends Immunol. 2013, 34, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Adiko, A.C.; Babdor, J.; Gutierrez-Martinez, E.; Guermonprez, P.; Saveanu, L. Intracellular Transport Routes for MHC I and Their Relevance for Antigen Cross-Presentation. Front. Immunol. 2015, 6, 335. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Ripoche, A.C.; Matheoud, D.; Nascimbeni, M.; Escriou, N.; Lebon, P.; Heshmati, F.; Guillet, J.G.; Gannage, M.; Caillat-Zucman, S.; et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity 2007, 27, 481–492. [Google Scholar] [CrossRef]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sorensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klechevsky, E.; Flamar, A.L.; Cao, Y.; Blanck, J.P.; Liu, M.; O’Bar, A.; Agouna-Deciat, O.; Klucar, P.; Thompson-Snipes, L.; Zurawski, S.; et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 2010, 116, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Oberkampf, M.; Guillerey, C.; Mouries, J.; Rosenbaum, P.; Fayolle, C.; Bobard, A.; Savina, A.; Ogier-Denis, E.; Enninga, J.; Amigorena, S.; et al. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos-Amador, P.; Perez-Cabezas, B.; Izquierdo-Useros, N.; Puertas, M.C.; Martinez-Picado, J.; Pujol-Borrell, R.; Naranjo-Gomez, M.; Borras, F.E. Capture of cell-derived microvesicles (exosomes and apoptotic bodies) by human plasmacytoid dendritic cells. J. Leukoc. Biol. 2012, 91, 751–758. [Google Scholar] [CrossRef]
- Bracamonte-Baran, W.; Florentin, J.; Zhou, Y.; Jankowska-Gan, E.; Haynes, W.J.; Zhong, W.; Brennan, T.V.; Dutta, P.; Claas, F.H.J.; van Rood, J.J.; et al. Modification of host dendritic cells by microchimerism-derived extracellular vesicles generates split tolerance. Proc. Natl. Acad. Sci. USA 2017, 114, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Salvi, V.; Gianello, V.; Busatto, S.; Bergese, P.; Andreoli, L.; D’Oro, U.; Zingoni, A.; Tincani, A.; Sozzani, S.; Bosisio, D. Exosome-delivered microRNAs promote IFN-alpha secretion by human plasmacytoid DCs via TLR7. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Loschko, J.; Heink, S.; Hackl, D.; Dudziak, D.; Reindl, W.; Korn, T.; Krug, A.B. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J. Immunol. 2011, 187, 6346–6356. [Google Scholar] [CrossRef]
- Loschko, J.; Schlitzer, A.; Dudziak, D.; Drexler, I.; Sandholzer, N.; Bourquin, C.; Reindl, W.; Krug, A.B. Antigen delivery to plasmacytoid dendritic cells via BST2 induces protective T cell-mediated immunity. J. Immunol. 2011, 186, 6718–6725. [Google Scholar] [CrossRef]
- Dubsky, P.; Saito, H.; Leogier, M.; Dantin, C.; Connolly, J.E.; Banchereau, J.; Palucka, A.K. IL-15-induced human DC efficiently prime melanoma-specific naive CD8+ T cells to differentiate into CTL. Eur. J. Immunol. 2007, 37, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Murata, K.; Fukushima, T.; Sugahara, K.; Tsuruda, K.; Anami, M.; Onimaru, Y.; Tsukasaki, K.; Tomonaga, M.; Moriuchi, R.; et al. A novel plasmacytoid dendritic cell line, CAL-1, established from a patient with blastic natural killer cell lymphoma. Int. J. Hematol. 2005, 81, 148–154. [Google Scholar] [CrossRef]
- Narita, M.; Watanabe, N.; Yamahira, A.; Hashimoto, S.; Tochiki, N.; Saitoh, A.; Kaji, M.; Nakamura, T.; Furukawa, T.; Toba, K.; et al. A leukemic plasmacytoid dendritic cell line, PMDC05, with the ability to secrete IFN-alpha by stimulation via Toll-like receptors and present antigens to naive T cells. Leuk. Res. 2009, 33, 1224–1232. [Google Scholar] [CrossRef]
- Drexler, H.G.; Macleod, R.A. Malignant hematopoietic cell lines: In vitro models for the study of plasmacytoid dendritic cell leukemia. Leuk. Res. 2009, 33, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Schuette, V.; Semmling, V.; Hochheiser, K.; Lukacs-Kornek, V.; Knolle, P.A.; Kurts, C. Steady-state cross-presentation of OVA is mannose receptor-dependent but inhibitable by collagen fragments. Proc. Natl. Acad. Sci. USA 2010, 107, E48–E49, author reply E50-41. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Yao, Y.; Zhou, L.; Mi, Q.-S.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. pDC-derived exosomes employ a novel mechanism to induce CD8 T cell responses in vivo. Center for Cutaneous Biology and Immunology, Department of Dermatology, Henry Ford Health System, Detroit, MI, USA, 2021. manuscript in preparation.
- Yao, Y.; Subedi, K.; Sexton, J.Z.; Liu, T.; Khalasawi, N.; Pretto, C.D.; Wotring, J.W.; Wang, J.; Yin, C.; Jiang, A.; et al. Circulating monocytes co-expressing surface ACE2 and TMPRSS2 upon TLR4/7/8 activation are susceptible to SARS-CoV-2 infection. Cell Res. 2021, submitted. [Google Scholar]
- Heine, A.; Juranek, S.; Brossart, P. Clinical and immunological effects of mRNA vaccines in malignant diseases. Mol. Cancer 2021, 20, 52. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Gebre, M.S.; Brito, L.A.; Tostanoski, L.H.; Edwards, D.K.; Carfi, A.; Barouch, D.H. Novel approaches for vaccine development. Cell 2021, 184, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Vannberg, F.O.; Dixon, J.B. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci. Rep. 2016, 6, 24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Phase | Exosomes /Antigen | Doses | Patients | Toxicity | Clinical Outcomes |
|---|---|---|---|---|---|---|
| Advanced Non-small cell lung cancer | I | Exosomes were isolated from autologous MoDCs generated in vitro, and loaded with MAGE peptides | once weekly for 4 weeks | 13 (9 completed) HLA-A2+ stage IIIb and IV NSCLC patients with tumor expression of MAGE3 or MAGE4 | Grade 1–2 toxicity | DTH reactivity against MAGE peptides in 3/9; MAGE-specific T cell responses in 1/3 patients examined; increased NK lytic activity in 2/4 [79]. |
| MAGE3- expressing advanced melanoma | I | Autologous MoDC-derived exosomes were loaded with MAGE3 peptides | once weekly for 4 weeks | 15 stage IIIb and IV, HLA-A1+, B35+ or HLA-DPO4+ patients | Only grade 1 toxicity | No detectable MAGE3-specific CD4 and CD8 T cell responses; restored NKG2D expression and NKG2D-dependent function of NK cells in 7/14 patients; 1/15 partial responses [67,80]. |
| Advanced colorectal cancer | I | Exosomes from patient ascites + GM-CSF, ASexos contained CEA with no additional antigen loading. | once weekly for 4 weeks | 40 HLA-A2+CEA+ stage III and IV CRC patients | Grade 1–2 toxicity | DTH induction in both groups, and CEA-specific CTL responses were observed in ASexo + GM-CSF group. 1 stable disease and 1 minor response in ASexo + GM-CSF group [82]. |
| Non-small cell lung cancer | II | IFN-γ-matured autologous MoDCs were loaded with both MHCI and MHCII tumor epitopes. | exosome immunization in 1, 2 and 3 week intervals in a maintenance immunotherapy regime | 26 (22 HLA-A2+ stage IIIb and IV NSCLC patients | 1/22 grade 3 hepato-toxicity | No detectable induction of antigen-specific T cell responses; increased NKp30-dependent NK cell function; 7 patients (32%) with progression-free survival at 4 months after chemotherapy cessation; no objective tumor response according to RECIST criteria [81]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.; Fu, C.; Zhou, L.; Mi, Q.-S.; Jiang, A. DC-Derived Exosomes for Cancer Immunotherapy. Cancers 2021, 13, 3667. https://doi.org/10.3390/cancers13153667
Yao Y, Fu C, Zhou L, Mi Q-S, Jiang A. DC-Derived Exosomes for Cancer Immunotherapy. Cancers. 2021; 13(15):3667. https://doi.org/10.3390/cancers13153667
Chicago/Turabian StyleYao, Yi, Chunmei Fu, Li Zhou, Qing-Sheng Mi, and Aimin Jiang. 2021. "DC-Derived Exosomes for Cancer Immunotherapy" Cancers 13, no. 15: 3667. https://doi.org/10.3390/cancers13153667
APA StyleYao, Y., Fu, C., Zhou, L., Mi, Q.-S., & Jiang, A. (2021). DC-Derived Exosomes for Cancer Immunotherapy. Cancers, 13(15), 3667. https://doi.org/10.3390/cancers13153667