Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression


 , , , , ,
, , , , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Melanoma Cell Lines
2.2. Immunoblotting
2.3. Mouse Studies
2.4. Cytoplasmic DNA Immunofluorescence
2.5. Immunohistochemistry
2.6. NanoString Analysis
2.7. Flow Cytometry
2.7.1. ICD Markers
2.7.2. Immune Profiling
2.7.3. Polyfunctional T Cell Assay
2.7.4. Cytokine Bead Array
2.8. ELISpot Assay of Mouse Splenocytes
3. Results
3.1. CHK1i+LDHU Triggers Pro-Inflammatory Cytokine Expression Independent of cGAS-STING and Immunogenic Cell Death
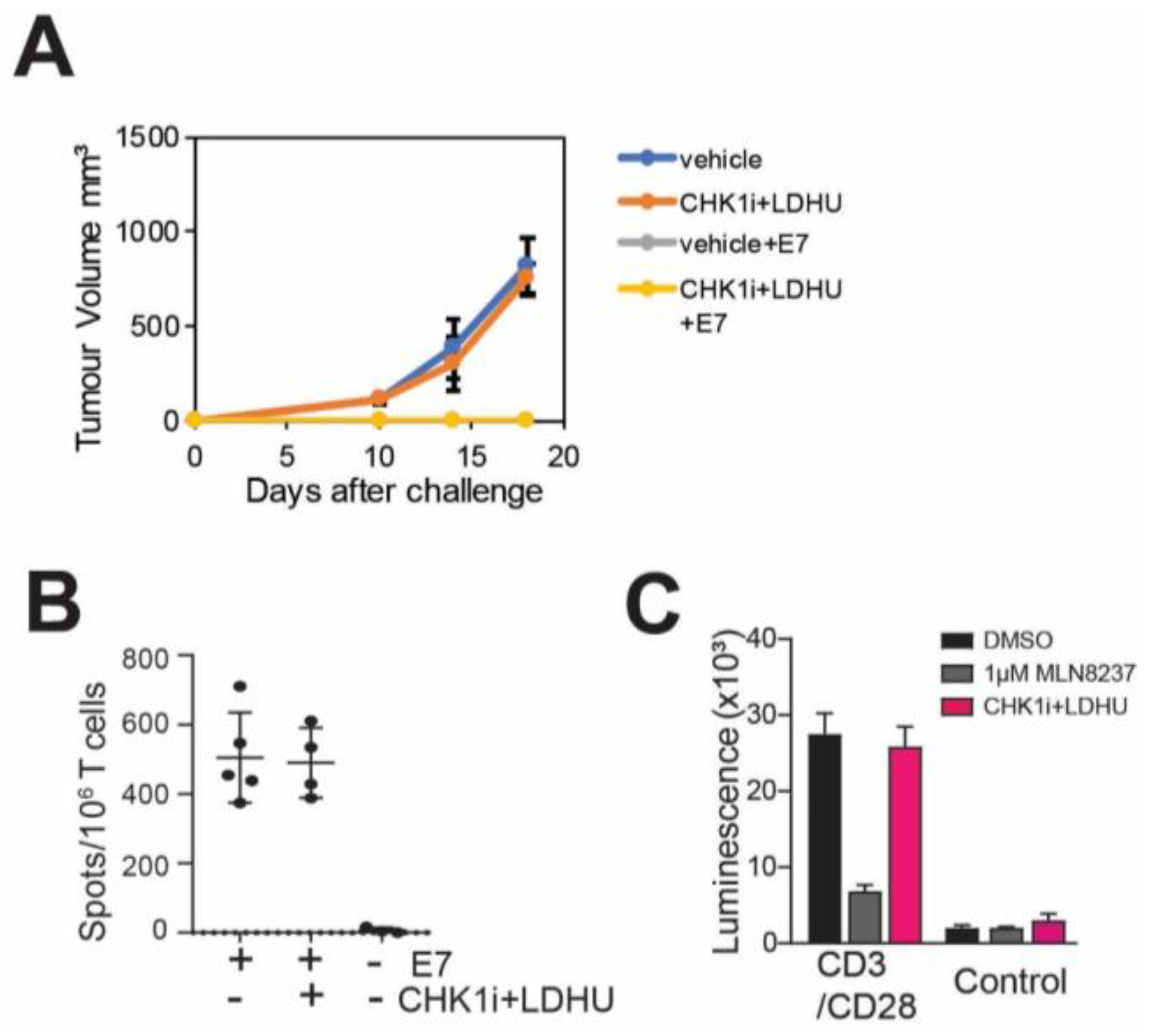
3.2. CHK1i+LDHU Does Not Adversely Affect T-Cell Proliferation or a T-Cell Mediated Immune Response
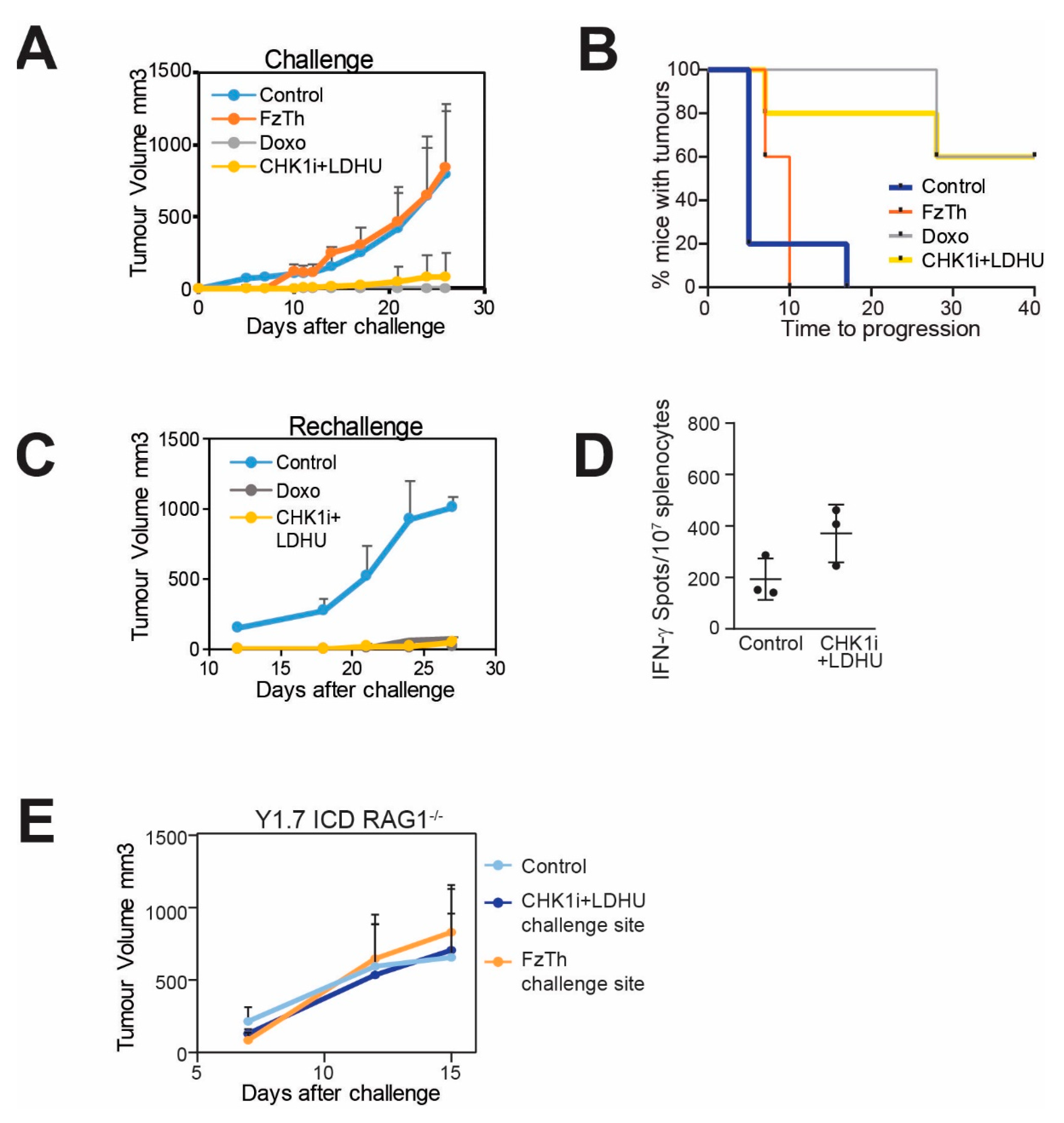
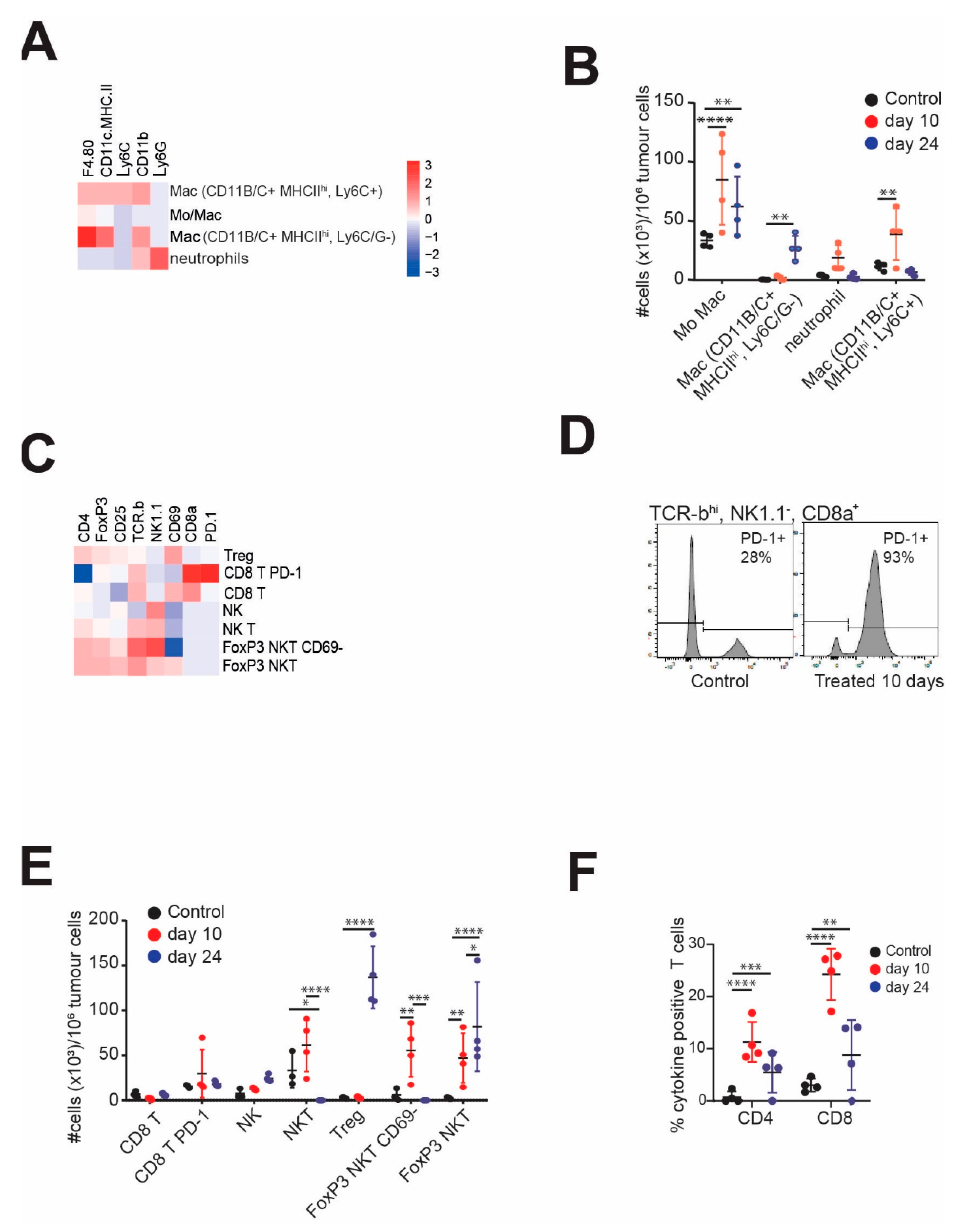
3.3. CHK1i+LDHU Triggers an Active Immune Response In Vivo
3.4. Efficacy of the CHK1i+LDHU Combined with Immune Checkpoint Blockade
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Lau, P.K.; Ascierto, P.A.; McArthur, G. Melanoma: The intersection of molecular targeted therapy and immune checkpoint inhibition. Curr. Opin. Immunol. 2016, 39, 30–38. [Google Scholar] [CrossRef]
- Twitty, C.G.; Huppert, L.A.; Daud, A.I. Prognostic biomarkers for melanoma immunotherapy. Curr. Oncol. Rep. 2020, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Rutkowski, P.; Hassel, J.C.; McNeil, C.M.; Kalinka, E.A.; et al. Five-year outcomes with nivolumab in patients with wild-type BRAF advanced melanoma. J. Clin. Oncol. 2020, 38, 3937–3946. [Google Scholar] [CrossRef]
- Chicas-Sett, R.; Morales-Orue, I.; Rodriguez-Abreu, D.; Lara-Jimenez, P. Combining radiotherapy and ipilimumab induces clinically relevant radiation-induced abscopal effects in metastatic melanoma patients: A systematic review. Clin. Transl. Radiat. Oncol. 2018, 9, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef]
- Cook, A.M.; Lesterhuis, W.J.; Nowak, A.K.; Lake, R.A. Chemotherapy and immunotherapy: Mapping the road ahead. Curr. Opin. Immunol. 2016, 39, 23–29. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Oo, Z.Y.; Stevenson, A.J.; Proctor, M.; Daignault, S.M.; Walpole, S.; Lanagan, C.; Chen, J.; Skalamera, D.; Spoerri, L.; Ainger, S.A.; et al. Endogenous replication stress marks melanomas sensitive to CHEK1 inhibitors in vivo. Clin. Cancer Res. 2018, 24, 2901–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Nazareth, D.; Jones, M.J.; Gabrielli, B. Everything in moderation: Lessons learned by exploiting moderate replication stress in cancer. Cancers 2019, 11, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Calvo, E.; Braiteh, F.; Von Hoff, D.; McWilliams, R.; Becerra, C.; Galsky, M.D.; Jameson, G.; Lin, J.; McKane, S.; Wickremsinhe, E.R.; et al. Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors. Oncology 2016, 91, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Oo, Z.Y.; Proctor, M.; Stevenson, A.J.; Nazareth, D.; Fernando, M.; Daignault, S.M.; Lanagan, C.; Walpole, S.; Bonazzi, V.; Skalamera, D.; et al. Combined use of subclinical hydroxyurea and CHK1 inhibitor effectively controls melanoma and lung cancer progression, with reduced normal tissue toxicity compared to gemcitabine. Mol. Oncol. 2019, 13, 1503–1518. [Google Scholar] [CrossRef] [Green Version]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Meeth, K.; Wang, J.X.; Micevic, G.; Damsky, W.; Bosenberg, M.W. The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. 2016, 29, 590–597. [Google Scholar] [CrossRef]
- Brooks, K.; Oakes, V.; Edwards, B.; Ranall, M.; Leo, P.; Pavey, S.; Pinder, A.; Beamish, H.; Mukhopadhyay, P.; Lambie, D.; et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013, 32, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Škalamera, D.; Stevenson, A.J.; Ehmann, A.; Ainger, S.A.; Lanagan, C.; Sturm, R.A.; Gabrielli, B. Melanoma mutations modify melanocyte dynamics in co-culture with keratinocytes or fibroblasts. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Chandra, J.; Dutton, J.L.; Li, B.; Woo, W.-P.; Xu, Y.; Tolley, L.K.; Yong, M.; Wells, J.W.; Leggatt, R.G.; Finlayson, N.; et al. DNA vaccine encoding HPV16 oncogenes E6 and E7 induces potent cell-mediated and humoral immunity which protects in tumor challenge and drives E7-expressing skin graft rejection. J. Immunother. 2017, 40, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination treatment of the oral CHK1 inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Tindle, R.W.; Fernando, G.J.; Sterling, J.C.; Frazer, I.H. A “public” T-helper epitope of the E7 transforming protein of human papillomavirus 16 provides cognate help for several E7 B-cell epitopes from cervical cancer-associated human papillomavirus genotypes. Proc. Natl. Acad. Sci. USA 1991, 88, 5887–5891. [Google Scholar] [CrossRef] [Green Version]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: A foe or ally? Cell. Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cai, Y.; Liu, L.; Wu, Y.; Xiong, X. Crucial biological functions of CCL7 in cancer. PeerJ 2018, 6, 4928. [Google Scholar] [CrossRef]
- Fankhauser, M.; Broggi, M.A.S.; Potin, L.; Bordry, N.; Jeanbart, L.; Lund, A.W.; Da Costa, E.; Hauert, S.; Rincon-Restrepo, M.; Tremblay, C.; et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving natural killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, 1452583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, T.; Lin, J.; Fatkhutdinov, N.; Liu, P.; Somasundaram, R.; Herlyn, M.; Zhang, R.; Nishigori, C. ARID2 deficiency correlates with the response to immune checkpoint blockade in melanoma. J. Investig. Dermatol. 2020, 141, 1564–1572. [Google Scholar] [CrossRef]
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Nguyen, X.N.; Wang, L.; Appourchaux, R.; Zhang, C.; Panthu, B.; Gruffat, H.; Journo, C.; Alais, S.; Qin, J.; et al. The interferon stimulated gene 20 protein (ISG20) is an innate defense antiviral factor that discriminates self versus non-self translation. PLoS Pathog. 2019, 15, 1008093. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.D. ZBP1: Innate sensor regulating cell death and inflammation. Trends Immunol. 2018, 39, 123–134. [Google Scholar] [CrossRef]
- Monteiro, M.; Almeida, C.F.; Caridade, M.; Ribot, J.C.; Duarte, J.; Agua-Doce, A.; Wollenberg, I.; Silva-Santos, B.; Graca, L. Identification of regulatory Foxp3+ invariant NKT cells induced by TGF-beta. J. Immunol. 2010, 185, 2157–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA damage response and immune defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING signaling in melanoma cells shapes antigenicity and can promote antitumor T-cell activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.; Wayne, J.; Massey, A.J. Checkpoint kinase 1 (Chk1) inhibition fails to activate the stimulator of interferon genes (STING) innate immune signalling in a human coculture cancer system. Mol. Biomed. 2021, 2, 19. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Nair, S.; Dhodapkar, M.V. Natural killer T cells in cancer immunotherapy. Front. Immunol. 2017, 8, 1178. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases iNOS(+)CD206(−) M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Travis, M.A.; Sheppard, D. TGF-β Activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.Y.; Lin, Y.C.; Mahalingam, J.; Huang, C.T.; Chen, T.W.; Kang, C.W.; Peng, H.M.; Chu, Y.Y.; Chiang, J.M.; Dutta, A.; et al. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res. 2012, 72, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, 16303. [Google Scholar] [CrossRef] [Green Version]
- Ondondo, B.; Jones, E.; Godkin, A.; Gallimore, A. Home sweet home: The tumor microenvironment as a haven for regulatory T cells. Front. Immunol. 2013, 4, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicci, D.G.; Hammond, K.J.; Uldrich, A.P.; Baxter, A.G.; Smyth, M.J.; Godfrey, D.I. A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J. Exp. Med. 2002, 195, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proctor, M.; Gonzalez Cruz, J.L.; Daignault-Mill, S.M.; Veitch, M.; Zeng, B.; Ehmann, A.; Sabdia, M.; Snell, C.; Keane, C.; Dolcetti, R.; et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers 2021, 13, 3733. https://doi.org/10.3390/cancers13153733
Proctor M, Gonzalez Cruz JL, Daignault-Mill SM, Veitch M, Zeng B, Ehmann A, Sabdia M, Snell C, Keane C, Dolcetti R, et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers. 2021; 13(15):3733. https://doi.org/10.3390/cancers13153733
Chicago/Turabian StyleProctor, Martina, Jazmina L. Gonzalez Cruz, Sheena M. Daignault-Mill, Margaret Veitch, Bijun Zeng, Anna Ehmann, Muhammed Sabdia, Cameron Snell, Colm Keane, Riccardo Dolcetti, and et al. 2021. "Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression" Cancers 13, no. 15: 3733. https://doi.org/10.3390/cancers13153733
APA StyleProctor, M., Gonzalez Cruz, J. L., Daignault-Mill, S. M., Veitch, M., Zeng, B., Ehmann, A., Sabdia, M., Snell, C., Keane, C., Dolcetti, R., Haass, N. K., Wells, J. W., & Gabrielli, B. (2021). Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers, 13(15), 3733. https://doi.org/10.3390/cancers13153733