
Synthetic Lethality Screening Highlights Colorectal Cancer Vulnerability to Concomitant Blockade of NEDD8 and EGFR Pathways

 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
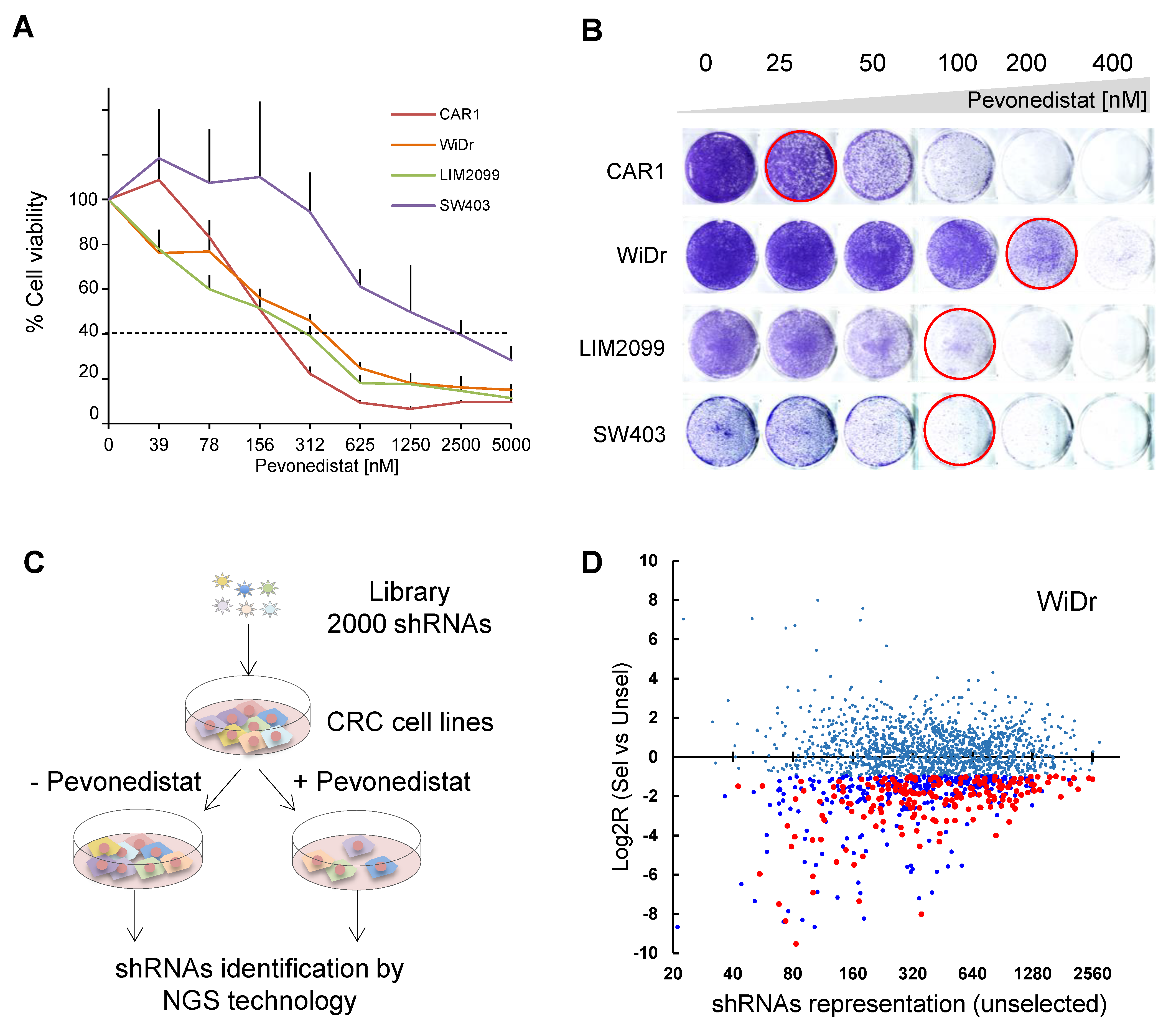
2.1. Genetic Screening for Synthetic Lethality with Pevonedistat in CRC Cell Lines
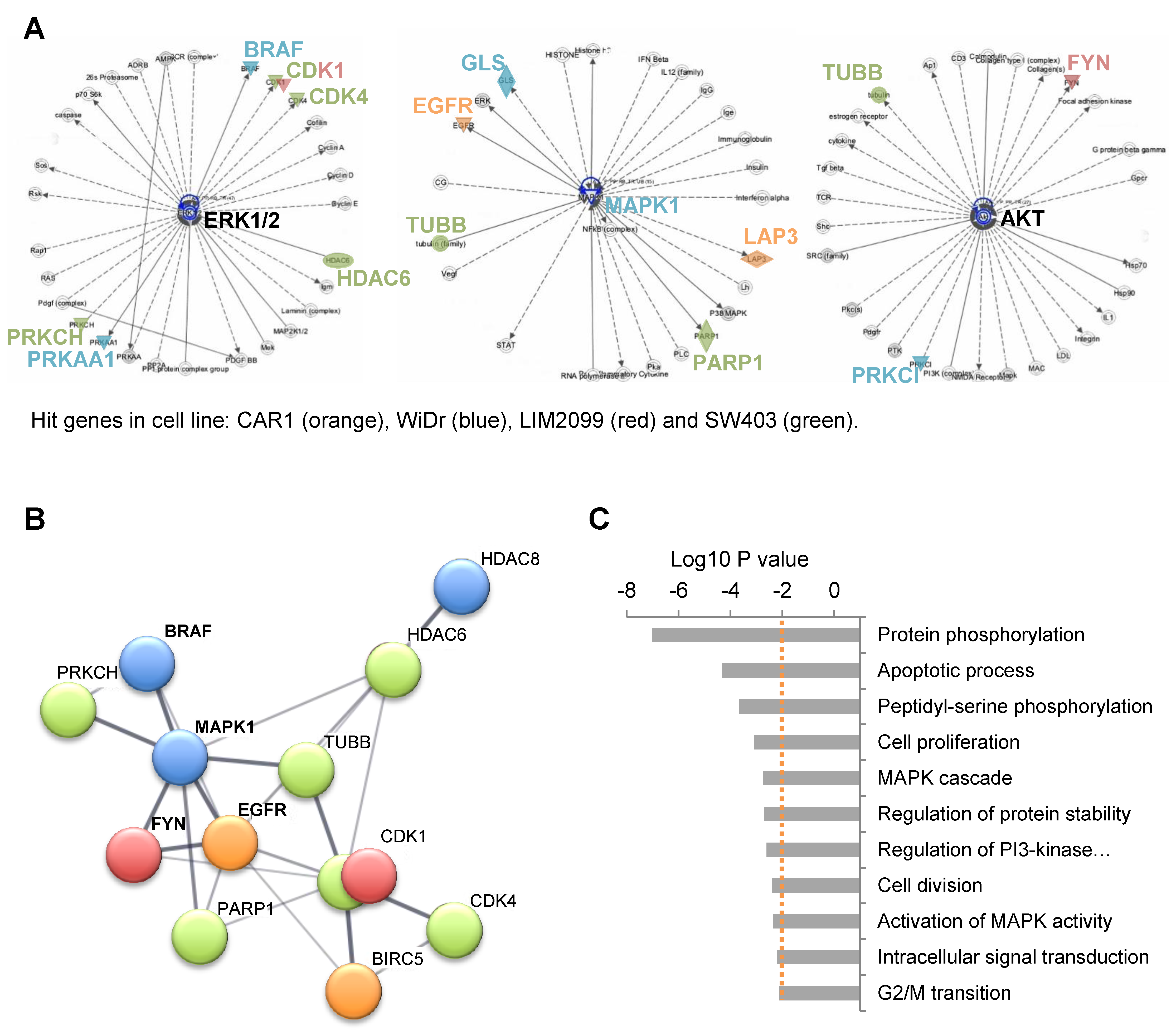
2.2. Involvement of EGFR Pathway Genes in Pevonedistat Synthetic Lethality
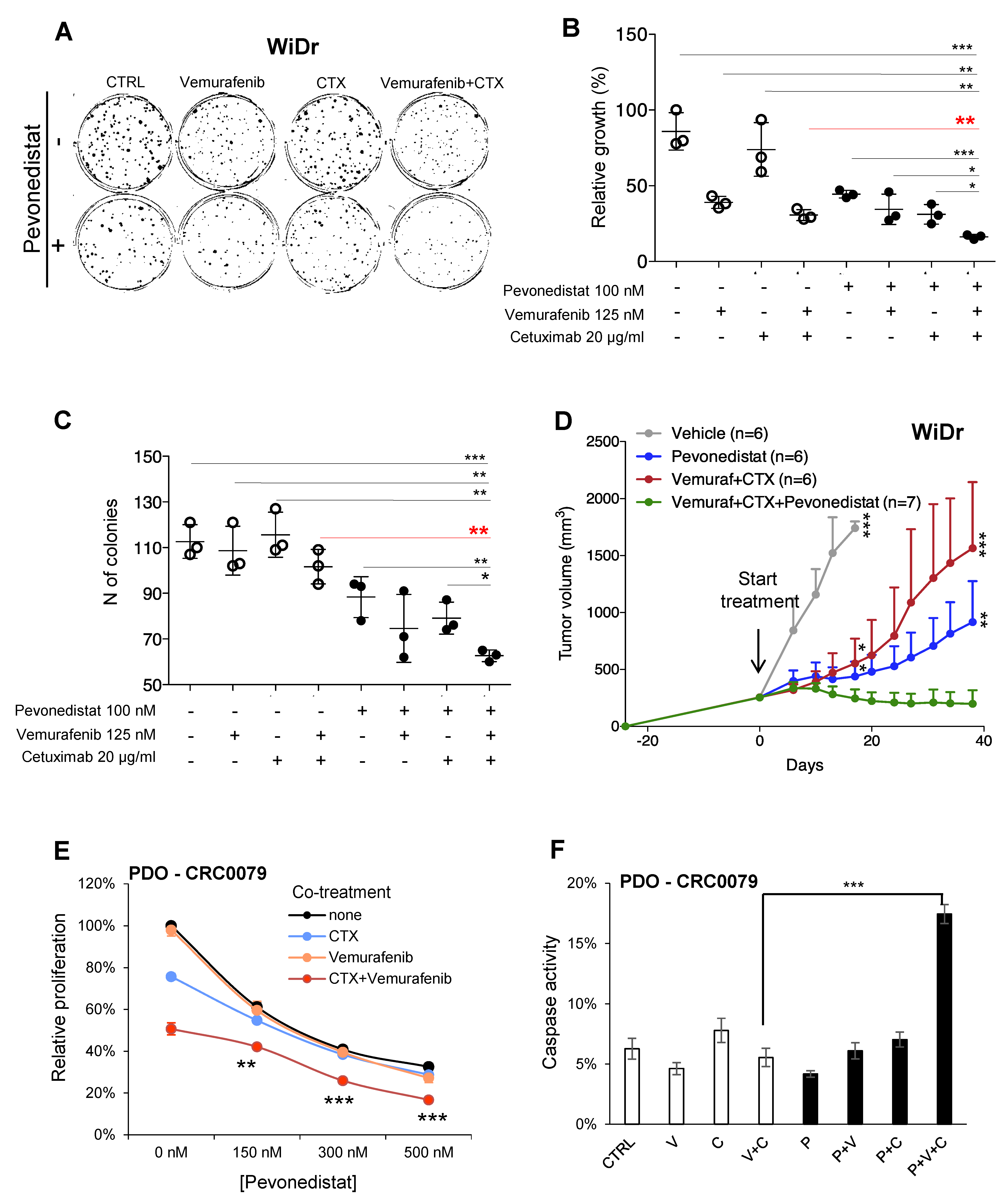
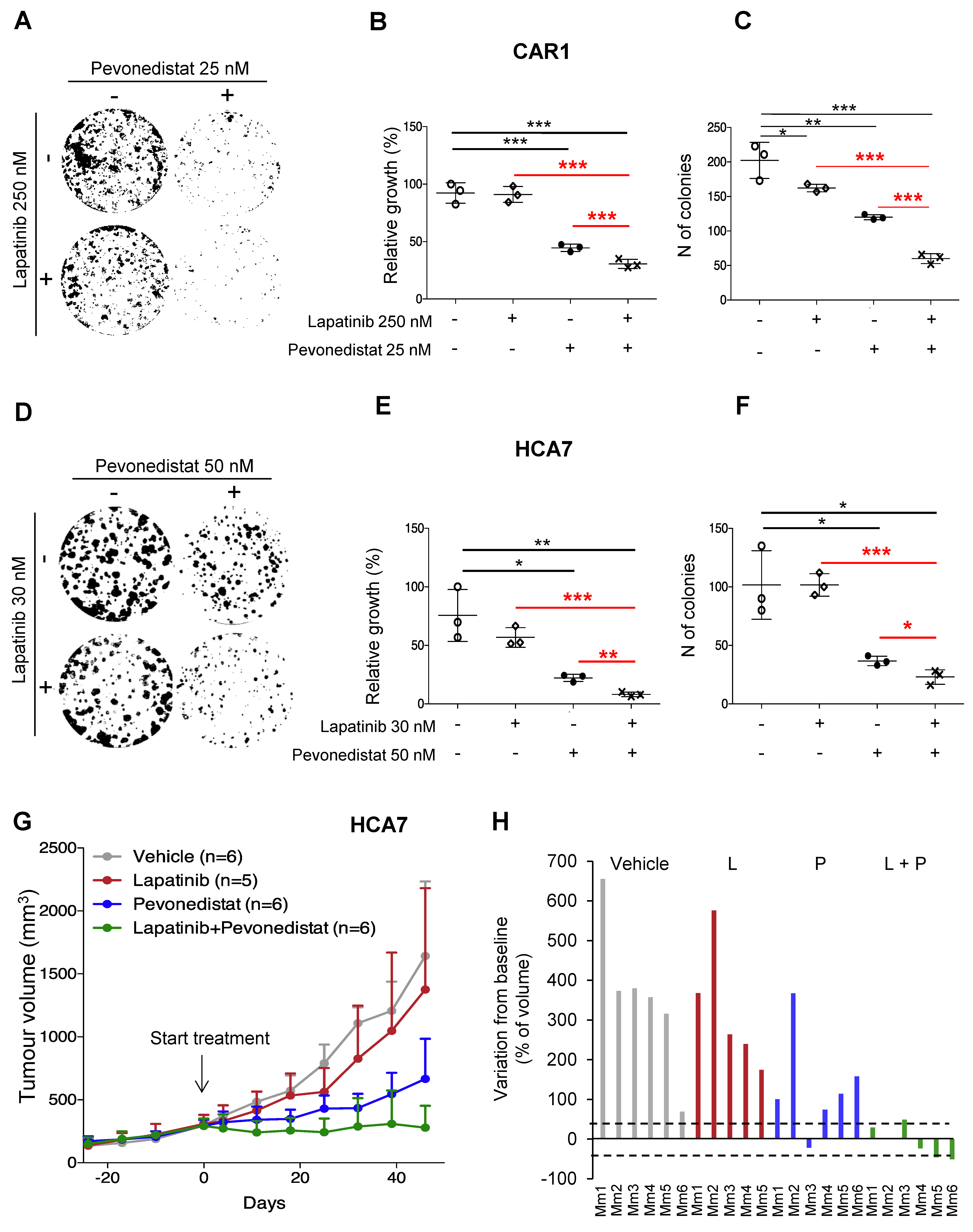
2.3. Pevonedistat Cooperates with BRAF and EGFR Inhibition in BRAF-Mutant CRCs
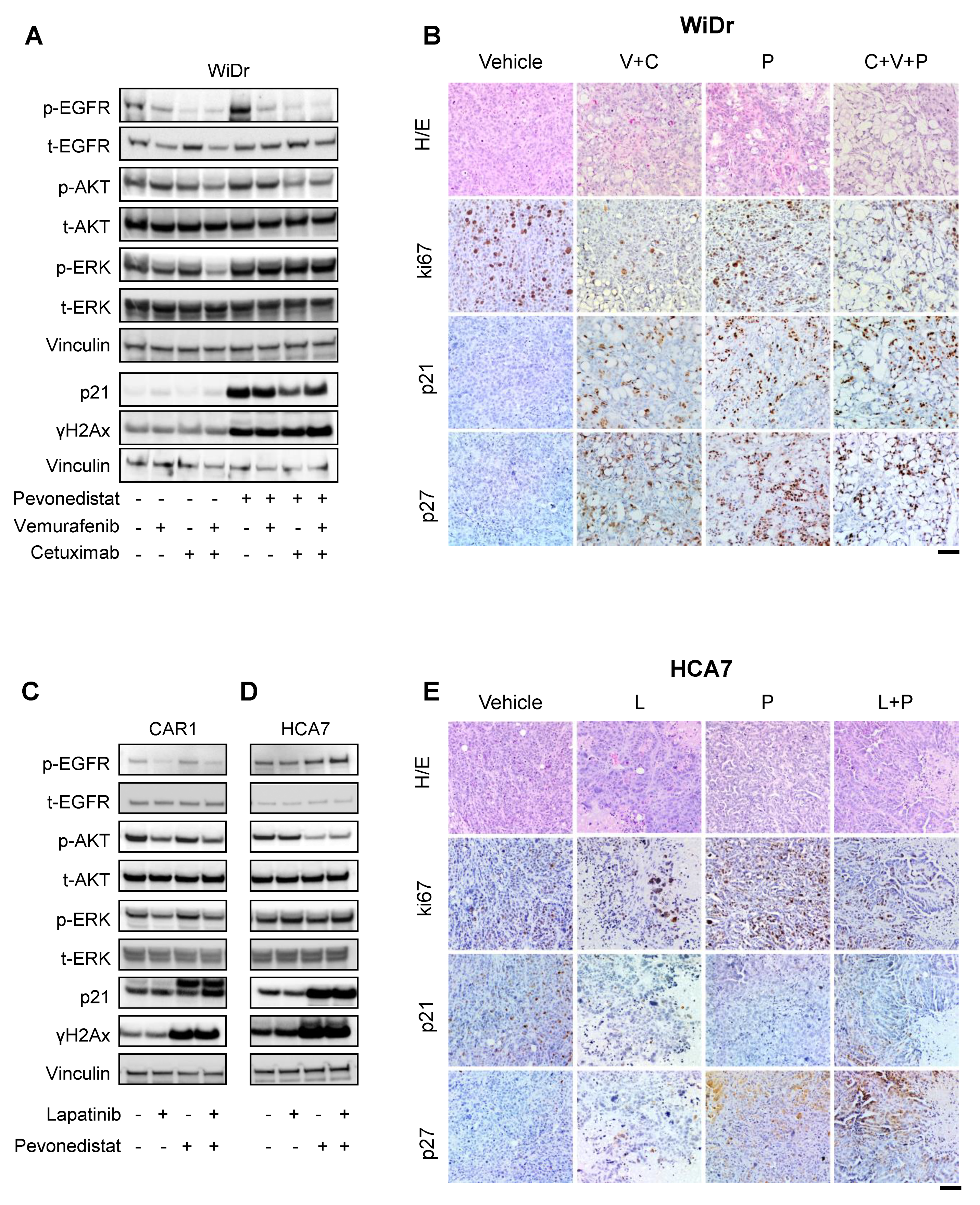
2.4. Combined Effect of NEDD8 and EGFR Pathway Blockade in RAS/RAF WT CRCs
2.5. Multiple Mechanisms of Cooperation between Pevonedistat and EGFR Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drugs
4.2. shRNA Library Design and Construction
4.3. Enrichment Analysis of Candidate Hits
4.4. In Vitro Drug Efficacy Studies
4.5. Organoids Drug Efficacy Study
4.6. Animals and Ethics Statement
4.7. In Vivo Studies
4.8. Immunoblot Analysis
4.9. Immunohistochemistry
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Mayer, R.J. Systemic therapy for colorectal cancer. N. Engl. J. Med. 2005, 352, 476–487. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O′Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Watson, I.R.; Irwin, M.S.; Ohh, M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 2011, 19, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Picco, G.; Petti, C.; Sassi, F.; Grillone, K.; Migliardi, G.; Rossi, T.; Isella, C.; Di Nicolantonio, F.; Sarotto, I.; Sapino, A.; et al. Efficacy of NEDD8 Pathway Inhibition in Preclinical Models of Poorly Differentiated, Clinically Aggressive Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Sarantopoulos, J.; Shapiro, G.I.; Cohen, R.B.; Clark, J.W.; Kauh, J.S.; Weiss, G.J.; Cleary, J.M.; Mahalingam, D.; Pickard, M.D.; Faessel, H.M.; et al. Phase I Study of the Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (TAK-924/MLN4924) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.J.; Jakubowiak, A.J.; O′Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Sekeres, M.A.; Fram, J.R.; Hua, Z.; Ades, L. Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J. Clin. Oncol. 2018, 36 (Suppl. 15). [Google Scholar] [CrossRef]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest. New Drugs 2019, 37, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Deeken, J.F.; Wang, H.; Subramaniam, D.; He, A.R.; Hwang, J.; Marshall, J.L.; Urso, C.E.; Wang, Y.; Ramos, C.; Steadman, K.; et al. A phase 1 study of cetuximab and lapatinib in patients with advanced solid tumor malignancies. Cancer 2015, 121, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Oved, S.; Mosesson, Y.; Zwang, Y.; Santonico, E.; Shtiegman, K.; Marmor, M.D.; Kochupurakkal, B.S.; Katz, M.; Lavi, S.; Cesareni, G.; et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J. Biol. Chem. 2006, 281, 21640–21651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milhollen, M.A.; Narayanan, U.; Soucy, T.A.; Veiby, P.O.; Smith, P.G.; Amidon, B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011, 71, 3042–3051. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Benamar, M.; Guessous, F.; Du, K.; Corbett, P.; Obeid, J.; Gioeli, D.; Slingluff, C.L.; Abbas, T. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine 2016, 10, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Li, H.; Sun, Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 2011, 13, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Leclerc, G.M.; Li, B.; Swords, R.T.; Barredo, J.C. Inhibition of the NEDD8 conjugation pathway induces calcium-dependent compensatory activation of the pro-survival MEK/ERK pathway in acute lymphoblastic leukemia. Oncotarget 2018, 9, 5529–5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Tan, M.; Nyati, M.K.; Zhao, Y.; Wang, G.; Sun, Y. Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E2935–E2944. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Shin, H.; Oh, J.W.; Oh, Y.J.; Her, N.G.; Nam, D.H. The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling. Cancers 2019, 11, 1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Kaufman, J.L.; Bernal, L.; Torre, C.; Matulis, S.M.; Harvey, R.D.; Chen, J.; Sun, S.Y.; Boise, L.H.; Lonial, S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 2014, 123, 3269–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers 2019, 11, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutational Status | CAR1 | WiDr | LIM2099 | SW403 |
|---|---|---|---|---|
| WT | BRAFm | KRASm | KRASm | |
| Candidate Genes | 3 | 6 | 2 | 7 |
| BIRC5 | + | − | − | − |
| EGFR | + | − | − | − |
| LAP3 | + | − | − | − |
| BRAF | − | + | − | − |
| GLS | − | + | − | − |
| HDAC8 | − | + | − | − |
| MAPK1 | − | + | − | − |
| PRKAA1 | − | + | − | − |
| PRKCI | − | + | − | − |
| CDK1 | − | − | + | + |
| FYN | − | − | + | − |
| CDK4 | − | − | − | + |
| HDAC6 | − | − | − | + |
| PARP1 | − | − | − | + |
| PGD | − | − | − | + |
| PRKCH | − | − | − | + |
| TUBB | − | − | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Invrea, F.; Punzi, S.; Petti, C.; Minelli, R.; Peoples, M.D.; Bristow, C.A.; Vurchio, V.; Corrado, A.; Bragoni, A.; Marchiò, C.; et al. Synthetic Lethality Screening Highlights Colorectal Cancer Vulnerability to Concomitant Blockade of NEDD8 and EGFR Pathways. Cancers 2021, 13, 3805. https://doi.org/10.3390/cancers13153805
Invrea F, Punzi S, Petti C, Minelli R, Peoples MD, Bristow CA, Vurchio V, Corrado A, Bragoni A, Marchiò C, et al. Synthetic Lethality Screening Highlights Colorectal Cancer Vulnerability to Concomitant Blockade of NEDD8 and EGFR Pathways. Cancers. 2021; 13(15):3805. https://doi.org/10.3390/cancers13153805
Chicago/Turabian StyleInvrea, Federica, Simona Punzi, Consalvo Petti, Rosalba Minelli, Michael D. Peoples, Christopher A. Bristow, Valentina Vurchio, Alessia Corrado, Alberto Bragoni, Caterina Marchiò, and et al. 2021. "Synthetic Lethality Screening Highlights Colorectal Cancer Vulnerability to Concomitant Blockade of NEDD8 and EGFR Pathways" Cancers 13, no. 15: 3805. https://doi.org/10.3390/cancers13153805
APA StyleInvrea, F., Punzi, S., Petti, C., Minelli, R., Peoples, M. D., Bristow, C. A., Vurchio, V., Corrado, A., Bragoni, A., Marchiò, C., Bertotti, A., Trusolino, L., Bardelli, A., Isella, C., Carugo, A., Draetta, G. F., & Medico, E. (2021). Synthetic Lethality Screening Highlights Colorectal Cancer Vulnerability to Concomitant Blockade of NEDD8 and EGFR Pathways. Cancers, 13(15), 3805. https://doi.org/10.3390/cancers13153805